
Chimie générale 15
Notions de thermodynamique chimique
1) Introduction
OBJET ET DOMAINE DE LA THERMODYNAMIQUE
Dans toute transformation de la matière , qu'elle soit physique ( fusion, ébullition... ) ou chimique ( réaction il y a deux niveaux de réalité auxquels correspondent deux niveaux de description.
- une réalité non-observable de manière directe , qui est celle des atomes et des molécules , de leurs interactions et de leurs comportements , et des changements intervenant dans leur organisation ( niveau microscopique )
- une réalité observable qui est celle des propriétés de la matière et des paramètres physiques caractéristiques de son état : pression, volume, température, conductivité électrique ;: production de chaleur ou de travail, changement de nature chimique , etc ( niveau macroscopique ).
La thermodynamique s'intéresse exclusivement au second aspect . C'est une science phénoménologique . Elle est indépendante de toute hypothèse ou de tout modèle particulier concernant la structure de la matière ; elle a d'ailleurs été fondée pour l'essentiel dans les premières décennies du XIX ème siècle , alors que les théories sur la structure de la matière commençaient seulement à s'ébaucher.
L'objet de la thermodynamique est de prévoir l'évolution d'un système dans des conditions données : peut-il évoluer ? Quelle est l'évolution possible ? Quel sera son aboutissement ?
Une construction "autoporteuse" reposant sur deux bases.
La thermodynamique repose sur deux notions : l'énergie et l'entropie , en affirmant que, dans l'univers que, la première se conserve et la seconde ne peut qu'augmenter. Ces deux affirmations sont des principes qui ne se démontrent pas , si ce n'est par la cohérence et l'exactitude des conséquence que l'on en tire.
Sur ces deux fondements, la thermodynamique a édifié une construction rigoureuse , d'une grande beauté formelle , qui n'obéit qu'à la logique mathématique . C'est donc une science par nature abstraite , dont les éléments essentiels sont des fonctions mathématiques et les relations qui les lient.
elle s'applique cependant, de façon très concrète , à de nombreux domaines : chimie et génie chimique , métallurgie, géologie , biologie, etc ; Les mêmes lois thermodynamiques s'appliquent à un réacteur industriel dans lequel a lieu une réaction , un alliage fondu, un nuage ou une cellule vivante.
La thermodynamique classique n'a besoin d'aucune hypothèse sur la structure de la matière , et se suffit à elle même. Mais n'est pas pour autant coupée du niveau corpusculaire et microscopique . Elle explique le comportement des systèmes ( par exemple la possibilité ou l'impossibilité d'une réaction spontanée et son résultat )en fonction des variations d'énergie et d'entropie , mais l'importance de ces variations est en définitive liée aux propriétés des atomes et des molécules , et aux changements de structure. Il se réalise donc , à ce niveau , une cohérence globale de la chimie.
La thermodynamique statistique , branche plus moderne de la thermodynamique , prend le parti d'appliquer une approche statistique à l'étude des systèmes considéréscomme des collections de particules.
Uniquement des bilans
Un trait caractéristique de la thermodynamique chimique est qu'elle se réfère uniquement aux états initial et final du système qui évolue, et au bilan des transformations . Le chemin suivi par la transformation peut avoir son importance ( réversibilité ), mais la thermodynamique ne cherche pas à élucider le mécanisme des transformations .Elle est donc radicalement différente , mais complémentaire , de la cinétique . Là ou celle-ci s'efforce de " reconstituer le film " de la transformation, la thermodynamique se contente de deux " phtographies " prises "avant" et "après" la transformation.
Le temps qui est au centre de la cinétique , n'apparaît donc pas en thermodynamique , si ce n'est implicitement et qualitativement. Parler d'un état final et d'un état initial suppose en effet qu'on les situe l'un par rapport à l'autre dans le temps , un temps qui ne se " remonte " pas.
2) Quelques rappels
L'atome gramme d'un élément est une quantité de cet élément égale à sa masse atomique exprimée en grammes.
La masse moléculaire d'un corps pur, simple ou composé, est égale à la somme des masse atomiques des atomes entrant dans la composition de la molécule de ce composé.
Une molécule gramme ou mole d'un corps pur est une quantité de ce corps égale à sa masse moléculaire exprimée en grammes. Un atome gramme de n'importe quel élément, une molécule gramme de n'importe quel corps pur contiennent le même nombre de particules ultimes, le nombre d'Avogadro : N = 6,02 . 1023.
L'équation chimique est une représentation qualitative et quantitative ( elle doit être équilibrée ) de la réaction chimique . Les corps du premier membre sont appelés réactifs, ceux du second produits de la réaction.
Certaines réactions chimiques sont complètes ( -------> ), d'autres sont limitées à un état d'équilibre chimique (<======>). Pour toute réaction chimique , il importe de préciser les conditions expérimentales : pression, température, solvant , pH du milieu.
Lors d'une réaction chimique, les lois stoechiométriques fondamentales ( Lavoisier, Proust, Dalton ) doivent être repectées.
2. Expressions de la composition d'un système chimique
Le système est une partie de l'univers, opposée au milieu extérieur . Le système peut être constitué d'une ou plusieurs phases ( partie homogène d'un système ). pour définir la composition d'un système et les modifications que celle-ci subit suite aux réactions chimiques, on utilisera les grandeurs suivantes :
- Fraction molaire d'un constituant = rapport du nombre de moles de ce constituant au nombre total de moles du systèmeou de la phase. La somme des fractions molaires est évidemment égale à l'unité.
- Molarité, ou concentration molaire CA ou [A] d'un composant = nombre de moles de composant par litre.
CA = [A] = nA/V
Par convention, pour les solutions diluées, on considère les concentrations entre crochets comme des concentrations réelles. Sinon on doit les multiplier par un coefficient d'activité inférieur à 1.
Les molarité sont notamment utiles dans les calculs de dilution : C1V1 = C2V2
Remarque : le volume variant avec la température , la concentration molaire est fonction de ce paramètre, alors que la fraction molaire en est indépendante.
- Concentration massique = nombre de grammes d'un composé par litre de solution.
molarité = concentration massique / masse moléculaire
pour les produits à l'état de trace ou d'impureté, la quantité est parfois exprimée en p.p.m ( parts par million ). 1 p.p.m = 1 mgr/kgr
- molalité = nombre de moles du constituant par kgr de solvant.
Elle est surtout utilisée pour des solutions très diluées. Contrairement à la molarité , elle est indépendante de la température.
- Normalité : capacité d'une solution d'échanger une mole' de particule ( N ) par litre
acidimétrie : 1 ion gr H+
oxydo-réduction : 1 électron gr
- Pression partielle d'un constituant d'un mélange gazeux = pression de ce gaz q'il occupait seul le volume offert du mélange : elle est donnée par :
pi. Vi = ni. R .T avec
p totale = ∑ pi
La pression totale d'un mélange gazeux est égale à la somme des pressions partielles des constituants.
3) quelques définitions importantes
- On peut définir un système comme " la partie de l'univers au sein de laquelle s'effectue la transformation étudiée". On oppose ainsi à la définition du système celle de l'environnement ou du milieu extérieur , c'est à dire tout le reste de l'univers avec lequel le système peut avoir des échanges de matières ou d'énergie.
Un système peut être " ouvert", " fermé" ou "isolé", et chacun de ces termes a un sens précis ;
- Système ouvert : il peut échanger avec l'extérieur de la matière et de l'énergie ( chaleur, travail mécanique ou électrique, rayonnement ). Exemples : un feu de bois , un réacteur ouvert à l'air libre et non isolé thermiquement.
- Système fermé : il peut échanger avec l'extérieur de l'énergie mais pas de la matière . Exemples : le circuit de fluide d'un réfrigérateur , l'ensemble cylindre / piston déjà décrit, qui peut être chauffé ou refroidit , et qui peut produire ou recevoir du travail par déplacement du piston.
- système isolé : il ne peut échanger avec l'extérieur ni énergie ni matière . Exemple : un réacteur clos, de volume invariable et isolé thermiquement . Il est en pratique de réaliser un système parfaitement isolé en empéchant totalement les les échanges d'énergie. On considère que l'univers , pris dans son ensemble , en constitue un , puisqu'il ne peut échanger de l'énergie avec rien.
La délimitation entre le " système" et " le milieu extérieur " est largement arbitraire , et le choix qui est fait peut modifier totalement les données d'un problème ( et rendre plus ou moins facile son étude ) . Ainsi lorsqu'on réalise une réaction dans une " bombe calorimétrique " immergée dans l'eau d'un calorimètre , la bombe constitue un système fermé mais non isolé , puisqu'elle fournit de la chaleur à son environnement , constitué par l'eau du calorimètre . Mais l'ensemble " bombe + calorimètre " constitue un système isolé ; la chaleur qui y est produite n'est pas échangée avec l'extérieur qui est alors constitué par le support deu calorimètre et par l'atmosphère.
4) Etat d'un système. Variables et fonctions d'état.
Une transformation est le passage d'un système d'un état dit initial à une autre dit final . Elle n'est définie si ces deux états sont eux-même définis , et ils le sont si on dispose à leur sujet des informations nécessaires pour pouvoir les reproduire identiquement.
Pratiquement, un état est défini par l'ensemble des valeurs d'un certain nombre de variables macroscopiques , appelées pour cette raison variables d'état . Pour un système physicochimique, ce sont par exemple la masse, la quantité de matière, le volume, la pression , la température, les concentrations , les pressions partielles , etc. Un mélange d'eau et de glace se trouve dans un état parfaitement défini si on connaît sa température , la pression qui s'exerce sur lui et la masse de chacune des deux phases.
Certaines de ces variables peuvent être liées entre elles par une relation appelée équation d'état, L'exemple le plus connu est celui du gaz parfait pour lequel P,V,n et T sont liés par la relation PV = nRT. Il suffit donc, pour définir l'état d'une certaine quantité n de gaz parfait, de connaître la valeur de deux des trois variables P,V et T, puisque celle de la troisième en résulte . On peut dire également que, pour cette quantité n de gaz parfait, on ne peut choisir librement que la valeur de deux des variables , celle de la troisième étant alors imposée.
- Variables extensives et variables intensives
Parmi les variables d'état, certaines sont proportionnelles à la quantité de matière ; on les appelle variables extensives . Exemples la masse, le volume. Elles sont additives lors de la réunion des deux systèmes de même nature , puisque la masse et le volume du nouveau système obtenu sont respectivement la somme des masses et des volumes des deux systèmes initiaux . Elles sont également multiplicatives : si l'on double la quantité de matière présente dans un système, sa masse et son volume doublent également.
d'autres variables sont indépendantes de la quantité de matière ; on les appelle variables intensives . Ce sont, par exemple la masse volumique, la concentration, la pression, la température, le potentiel chimique. Le potentiel chimique est la variable intensive associée à la variation de la quantité de matière ( moles ) d'un constituant, dans un système ou dans une phase, de même que, par exemple , la pression est la variable intensive associée aux variations du volume. La masse volumique du benzène est la même, qu'il s'agisse de 0,3 moles ou de 10 moles ; la concentration en sel de l'eau de mer est la même dans une goutte et dans un mètre-cube ( mais bien entendu, la quantité de sel - variable extensive - contenue dans une goutte d'eau et dans un mètre cube n'est pas la même . Les variables intensives ne sont pas additives lors de la réunion de deux systèmes : si on mélange de l'eau à 20 °C et de l'eau à 50°C, on obtient pas de l'eau à 70°C.
5. Fonctions d'état
Souvent, il est possible de réaliser une même transformation de plusieurs façons . Par exemple , on peut élever la température d'un morceau de métal en le mettant en contact avec une source de chaleur ou par frottement ( échauffement des freins )
Si les états initials et finals sont identiques , les variations des variables d'état au cours de cette transformation sont, par définition , les mêmes dans tous les cas. De même , la longitude, la latitude et l'altitude sont les variables d'étatqui définissent le point de départ ( état initial ) et le point d'arrivée ( état final ) du trajet d'une voiture . Leurs variations sont entièrement définies par les coordonnées de ces deux points , quelque soit l'itinéraire suivi.
Mais la thermodynamique définit aussi diverses grandeurs liées aux variables d'état ( chaleur ou travail échangé, énergie interne, etc )et l'on peut se demander si leurs variations , au cours d'une transformation , sont elles aussi indépendantes du "chemin suivi " . Celles qui satisfont cette condition sont appelées fonctions d'état . Elles ont une valeur définies pour chaque état du système et leurs variations sont donc entièrement définies par l'état initial et l'état final ( en appelant A et B ces deux états , et F la fonction d'état : ? FA-->B = (FB - FA) . Toutes ces grandeurs ne sont pas dans ce cas , de même que seule la variation d'énergie potentielle de la voiture ne dépend que des points de départ et d'arrivée , et non du parcours .
En langage mathématique : , une fonction d'état est une fonction F(x,y,z) de plusieurs variables d'état x,y et z ), caractérisées par l'existence d'une différentielle exacte ( on dit aussi différentielle totale exacte ) . Ceci signifie que l'accroissement dF de cette fonction qui accompagne des accroissements simultanés dx, dy, dz des variables est égal à la somme des accroissements de cette fonction accompagnant des accroissements séparés de chacune des variables , les autres restant constantes :
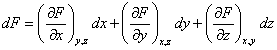
![]()
est la dérivée partielle de f par rapport à x,y et z étant constants .
6. Etats d'équilibre
Un système se trouve dans un état d'équilibre si les variables qui définissent son état ne variant pas au cours du temps , et si les variables intensives ont la même valeur dans tout l'étendue du système : par exemple, même température en tous points ( équilibre thermique ) à, même pression en tous les points ( équilibre mécanique ) .
On admet, d'une manière générale , que l'état initial et l'état final des transformations sont des états d'équilibres , sinon les variables d'état n'auraient pas une valeur initiale et une valeur finale définies, et leurs variations ne seraient pas non plus définie ( on verra cependant plus loin qu'il faut nuancer cette conception en ce qui concerne l'état initial ).
Un état d'équilibre constaté au niveau macroscopique n'exclut pas l'existence de transformation s'effectuant au niveau microscopique . Il s'agit alors d'un équilibre stationnaire , équilibre apparent résultant d'une compensation exacte entre les effets de deux transformations inverses l'une de l'autre s'effectuant à la même vitesse . Exemple : l'équilibre liquide-gaz ; les équilibres chimiques, comme l'estérification.
7. réversibilité thermodynamique
Une transformation est inversible si elle peut être réalisée dans les deux sens : le chauffage d'un corps et son refroidissement , la compression d'un gaz et son expansion, la fusion et la solidification , sont des exemples de transformations inverses . Les réactions chimiques sont, au moins en théorie toutes inversibles.
Le terme réversible s'applique à une transformation inversible réalisée en outre dans des conditions particulières. La réversibilité thermodynamique exige qu'au cours de la transformation le système passe par une infinité d'états intermédiaires différant peu d'états d'équilibres , et dont chacun diffère infiniment peu du suivant ( états " quasi statiques ). Dans ces conditions , les deux transformations inverses passent par les mêmes états intermédiaires , mais dans un ordre inverse.
Deux exemples illustreront et préciseront cette définition passablement abstraite :
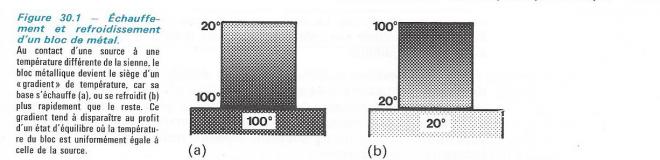
a) Pour porter un bloc de métal de 20°C à 100 °C on peut le poser sur une plaque maintenue à 100 °C. Son échauffement se fera progressivement à partir du bas et, jusqu'à ce qu'il soit entièrement à 100°C, sa température ne sera pas uniforme . Pour le ramener ensuite à 20°C, on peut le poser sur une autre plaque , maintenue à 20°C. Son refroidissement se fera aussi à partir du bas , jusqu'à ce qu'il soit entièrement revenu à 20°C, sa température ne sera encore pas uniforme .
Cette transformation est inversible , mais elle n'est pas effectuée ici de manière thermodynamiquement réversible . En effet, les états intermédiaires ne sont pas des états d'équilibre , puisque la température ( variable intensive ) n'est pas la même en tous les points du métal. En outre ils ne sont pas les mêmes dans les deux sens .
Pour la réaliser réversiblement , il faudrait mettre le bloc de métal en contact successivement avec une infinité de sources de chaleur dont les températures croissantes différeraient infiniment peu.
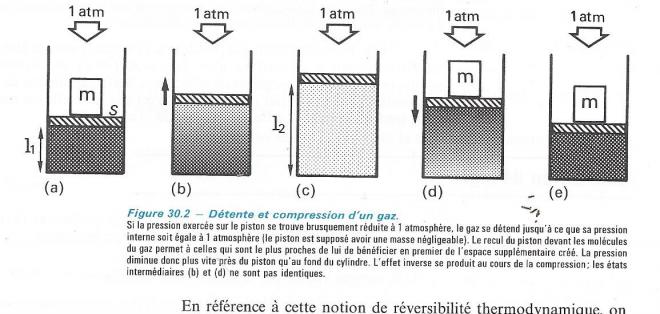
 b) Supposons qu'un gaz soit comprimé dans un cylindre par un piston sur lequel s'exerce, outre la pression atmosphérique , le poids d'une masse m , de telle sorte que la pression totale du gaz soit supérieure à la pression atmosphérique . Si l'on retire la masse m , le gaz se " détend" ; son volume augmente brutalement jusqu'à ce que la pression soit devenue égale à la pression atmosphérique . Si alors on replace la masse sur le piston , le gaz reprend toujours brusquement , son volume et sa pression de début.
b) Supposons qu'un gaz soit comprimé dans un cylindre par un piston sur lequel s'exerce, outre la pression atmosphérique , le poids d'une masse m , de telle sorte que la pression totale du gaz soit supérieure à la pression atmosphérique . Si l'on retire la masse m , le gaz se " détend" ; son volume augmente brutalement jusqu'à ce que la pression soit devenue égale à la pression atmosphérique . Si alors on replace la masse sur le piston , le gaz reprend toujours brusquement , son volume et sa pression de début.
En procédant ainsi, on ne réalise pas cette transformation dans des conditions de réversibilité . En effet, au cours de l'expansion comme au cours de la compression , la pression de gaz n'est pas uniforme dans tous le volume . La montée du piston provoque d'abord une décompression à son voisinage , de même que sa redescente comprime d'abord le gaz en contact avec lui. Les deux transformations ne passent en outre pas par les mêmes états intermédiaires.
La réversibilité exigerait que la pression exercée sur le gaz soit diminuée de façon infiniment progressive , de telle sorte qu'à chaque instant le gaz soit dans un état très voisin de l'équilibre ( pression uniforme ) . Pour s'approcher de la réversibilité , on pourrait , par exemple, fragmenter la masse m en n masses égales à M/n, que l'on retirerait l'une après l'autre ; mais pour atteindre les conditions de réversibilité il faudrait pouvoir fragmenter m en une infinité de masses infiniment petites . Une assez bonne approximation de la réversibilité pourrait être réalisée en utilisant comme masse m un liquide contenu dans un récipient , que l'on laisserait s'évaporer . Le départ de chaque molécule contribuerait à diminuer la pression exercée sur le gaz, mais de façon infime.
 En référence à cette notion de réversibilité thermodynamique , on peut distinguer deux types de transformations :
En référence à cette notion de réversibilité thermodynamique , on peut distinguer deux types de transformations :
- les transformations naturelles, qui ne s'effectuent jamais de façon strictement réversible, même si parfois elles s'en approche , par exemple en étant très lentes . l'état d'équilibre dans lequel se trouve initialement le gaz de l'exemple précédent résulte d'une contrainte imposée ( la force exercée par la masse m sur le piston ) La suppression de cette contrainte provoque une évolution spontanée non contrôlée , inversible mais non réversible , vers un autre état d'équilibre.
- les transformations guidées auxquelles on impose un déroulement différent du déroulement naturel ( par exemple en relâchant la pression exercée sur le gaz selon u " programme" dont on est maître) . Une transformation ne peut s'effectuier réversiblement que si elle est guidée , mais la réversibilité parfaite reste du domaine théorique ; une transformation réelle, même guidée, ne peut être parfaitement réversible.
8. Buts de la thermodynamique chimique
C'est une science qui étudie les échanges d'énergie survenant lors de toute transformation. Ces échanges révèlent des différences de stabilité entre produits et réactifs.
La thermodynamique étudie donc les chaleurs de réaction et permet de prévoir le sens des réactions.
9. Chaleurs de réaction
La chaleur est une forme d'énergie qui s'échange entre deux corps à des températures différentes . Elle peut s'exprimer dans toutes les unités d'énergie , mais nous n'utiliserons que le joule et la calorie ( 1 calorie = 4,18 joules ) .
Lors d'un processus chimique , on appelle chaleur de réaction la quantité de chaleur dégagée ou absorbée lors d'une réaction chimique où tous les réactifs sont transformés en produits ( transformation complète ).
Exemple :
C2H4 + H2 -----> C2H6
Chaleur de réaction = quantité de chaleur échangée lorsque 1 mole ( 28 gr. ) d'éthylène réagit avec 1 mole d'hydrogène ( 2 gr. ) pour donner 1 mole d'éthane ( 30 gr. ).
Convention de signe : tout gain de chaleur par le système est compté positivement, toute perte négativement, donc la chaleur de réaction est
positive si la réaction absorbe de la chaleur ( endothermique ).
négative si la réaction dégage de la chaleur ( exothermique ).
Les chaleurs de réaction se mesurent à l'aide d'un dispositif calorimétrique .
Cette chaleur de réaction est fonction de la différence de stabilité entre les réactifs et les produits, mais outre cette forme d'énergie échangée , il faut tenir compte notamment du travail des forces de pression.
Ainsi, supposons que la réaction ci dessus se produise à 0°C entre gaz à la pression atmosphérique . Le volume des réactifs est 2x 22,4 litres, celui du produit 22,4 litres. Le volume diminue , donc le système reçoit un travail effectué par la pression extérieure.
Au contraire, une réaction comme :
H2O (1) -------> H2(g) + 1/2 O2 (g)
se produit avec augmlentation de volume , le système fournit un travail contre les forces de pression.
La convention de signe est également valable pour le travail
Le travail reçu par le système est compté positivement
Le travail fourni par le système est compté négativement.
- Calcul du travail des forces de pression
Cas général
Soit un piston infiniment léger, mobile sans frottement dans un cylindre fermé de volume V, contenant une masse de gaz. Le système est formé par la masse de gaz et l'ensemble cylindre - piston. Le dépacement du piston suivant l'axe du cylindre, est positif lorsque le volume V croît. Supposons que le piston, suite à une réaction chimique dans le cylindre conduisant à une augmentation de volume , subisse un petit déplacement dx. Le système fournit alors un travail dW contre la pression extérieure , dont la force est Pext . S
Ce travail vaut dW = - pext . S . dx.
Travail fournit donc par convention négatif d'autant que pext, S, dx positifs ( travail fourni )
Or, S . dx = dV, variation du volume intérieur du cylindre donc dW = - pext . dV
en cas de déplacement dans l'autre sens, dx négatif, le travail serait positif (travail reçu).![]()
Pour un déplacement fini du piston, de V1 à V2, il suffit d'intégrer du volume initial au volume final :
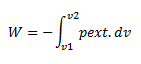

Cas particuliers
- si la pression extérieure est constante
( ex pext = pression atmosphérique )
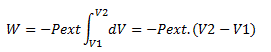
- travail réversible d'un gaz parfait à température constante
Transformation réversible : il y a à tout moment égalité entre entre pext et pint au cours de la transformation.
Pext = Pint
et : ![]()
Pour intégrer cette expression, exprimons Pint en fonction de V. Un gaz parfait obéit à la loi P.V=n.R.T
n = nombre de moles contenues dans le récipient.
Donc
![]()
et :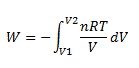
Comme la température reste constante :
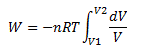
Ce qui donne par intégration :
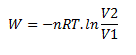 10. Premier principe de la Thermodynamique et applications
10. Premier principe de la Thermodynamique et applications
Le premier principe de la thermodynamique nous donne une relation entre les échanges de travail mécanique et de chaleur lors d'une réaction chimique
Enoncé : lors de l'évolution d'un système , la somme algébrique du travail et de la quantité de chaleur échangée avec l'extérieur , ne dépend que de l'état initial et l'état final.
Donc, pour passer d'un état A ( PA, TA, composition chimique donnée ) à l'état B ( PB, TB, autre composition chimique ), en échangeant avec le milieu extérieur de la chaleur et du travail, le système chimique peut emprunter n'importe quelle voie . Pour deux chemins quelconques 1 et 2, on aura toujours :
W1 + Q1 = W2 + Q2
ou encore W+Q = constante
 Si l'état initial est identique à l'état final :
Si l'état initial est identique à l'état final :
W + Q = 0
ou W = - Q
Donc dans le cas d'un cycle fermé , pour récupérer du travail, il faut donner de la chaleur et réciproquement
Pour utiliser plus commodément le premier principe , on pose que la somme W + Q est égale à la variation d'une fonction Un appelée énergie interne , qui ne dépend que de l'état initial A et de l'état final B ( fonction d'état ).
W + Q = UB - UA
ou W + Q =? U
Sens physique de l'énergie interne
D'après le premier principe, les variations d'énergie interne sont mesurables. Cette fonction représente en quelques sortes l'énergie que l'on peut extraire d'un système dans un état donné , énergie déterminée par :
- Les conditions physiques : P et T
- La position dans un champ ( de gravitation, électrique ou magnétique )
- Les liaisons entre atomes et molécules
- Les liaisons entre les électrons et le noyau atomique
- Les liaisons entre nucléons assurant la cohésion du noyau
- La masse des nucléons.
Cette énergie interne peut être évaluée en fonction de la relation d'Einstein : E = mc2
On imagine que sa valeur est infiniment grande vis à vis des variations que l'on mesure habituellement. On considèrera donc l'énergie interne d'un système comme un réservoir immense d'énergie dont on mesure seulement les variations de niveau.
L'énergie interne , proportionnelle à la quantité de matière est une grandeur extensive . La t° par exemple est au contraire une grandeur intensive
Cas particulier : système isolé ( n'échange avec l'environnement ni travail, ni chaleur ), donc
W = 0 Q = 0
et UB = UA
L'énergie interne d'un système isolé reste constante
L'univers peut être considéré comme un système isolé, son énergie interne reste constante
Pour définir l'état d'un système , on dispose des variables suivantes , pression, volume, température, composition chimique, nombre de phases. L'état d'un système ne peut être défini que par des variables indépendantes . Ainsi, on ne peut choisir ou fixer à la fois P,V et T puisque ces trois variables sont reliées entre-elles , qui est pour les gaz parfaits P.V = nRT.
Il y a donc seulement deux variables indépendantes , et, par commodité, on choisit souvent les deux variables intensives P et T
De même dans une réaction d'équilibre :
A + B <=====> C + D
Il est impossible de fixer les valeurs des fractions molaires des 4 corps, vu qu'elles sont reliées par la constante d'équilibre, Le nombre de variables indépendantes est donc de 3 et non de 4
Conditions pour que le chaleur de réaction soit variable d'état
Notre but est de déterminer les chaleurs de réaction à partir d'un minimum de résultats expérimentaux. Si elles étaient fonctions d'état, il suffirait d'une seule détermination suivant le chemin le plus facile à étudier expérimentalement , mais nous savons que c'est W + Q qui est variable d'état donc :
Q = UB - UA - W
dépend comme W du chemin parcouru.
Dans certaines conditions particulières , la chaleur de réaction est toutefois une fonction d'état
- Transformation à volume constant
Si le travail est nul, W = 0, on a Q = UB - UA et ne dépend donc que de l'état initial et de l'état final.
Or nous avons vu que : 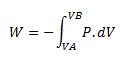 .
.
On ne peut supposer que p soit nulle , donc pour que W = 0 , il faut que dV = 0, donc que le volume reste constant.
Lorsqu'une réaction se produit à volume constant, la chaleur échangée est une fonction d'état.
On a alors : QV = UB - UA = ?U
QV = chaleur de réaction à volume constant
Si la réaction est exothermique, QV < 0 , UB < UA l'énergie interne du système décroît.
Si la réaction est endothermique, QV > 0 , UB > UA l'énergie interne du système croît.
Quand une réaction chimique s'effectue-t-elle à volume constant ?
- quand elle se produit dans une enceinte close et indéformable.
- réaction entre gaz, à t° constante , sans changement du nombre de môle .
- réactions en phase condensée entre liquides ou entre liquides et solides, car les variations de volume sont alors très faibles.
- Transformations à pression constante : définition de l'enthalpie.
Nous avons vu que, pour une transformation à P constante, le travail est donné par : W = - P . ( VB - VA ).
En remplaçant W par sa valeur dans W + Q = UB - UA , on obtient : - P . ( VB - VA ) + QP = UB - UA
QP = chaleur de réaction à pression constante
QP = UB - UA + P . ( VB - VA )
= ?U + P.?V
ou encore : QP = (UB + PVB) - (UA + PVA)
QP = (U + PV)B - (U + PV)A
La chaleur de réaction à pression constante ne dépend donc que de l'état initial et de l'état final , elle est égale à la variation d'une vonction d'état U + PV que l'on pose égale à H, appelée enthalpie
H = U + PV
On a donc : Qp = HB - HA = ?H
Donc pour une réaction exothermique , l'enthalpie diminue
pour une réaction endothermique , l'enthalpie croît
La variation d'enthalpie est égale à la variation d'énergie interne à pression constante corrigée du travail des forces de pression (*)
Le fait que la chaleur de réaction à pression constante soit une fonction d'état est intéressant, car de nombreuses réactions se produisent à la pression atmosphérique
(*) La majeure partie des expériences de chimie se déroulent dans des récipients ouverts à l'air libre et soumis à une pression constante, plutôt qu'à volume constant dans un récipient résistant et scellé. En général quand une transformation à lieu dans un système ouvert, le volume du système change . Par exemple la décomposition thermique de 1,0 mol de CaCO3 à 1 bar aboutit à un accroissement de volume de 89 L à 800°C à cause du dioxyde de carbone gazeux produit. Pour offrir ce grand volume au dioxyde de carbone, l'atmosphère environnante doit être repoussée . Cette augmentation de volume signifie que le système doit fournir un travail d'expansion . De ce fait, malgré l'apport de chaleur pour provoquer la décomposition endothermique , l'élévation d'énergie interne du système n'est pas égale à l'énergie fournie sous forme de chaleur parce qu'elle a en partie servi à effectuer le travail d'expansion. En d'autres termes , le volume ayant augmenté , une partie de la chaleur fournie au système est revenue dans l'environnement sous forme de travail. un autre exemple est l'oxydation d'un corps gras, comme la tristéarine , en dioxyde de carbone dans le corps. La réaction globale est :
2C57H110O6(aq) + 163 O2 (g) -------> 114CO2 + 110 H2O (l)
Dans cette réaction il y a une réduction nette de volume équivalente à l'élimination de 49 mol de molécules de gaz. La réduction de volume est d'environ 600 ml à 25 °C pour la consommation de 1g de cette graisse. Le volume occupé par les substances décroissant au cours de la réaction , l'atmosphère exerce une travail sur le système à mesure que la réaction se déroule . Cela veut dire qu'il y a transfert d'énergie sous forme de travail du milieu extérieur vers le système à mesure qu'il se contracte. De ce fait une plus grande quantité d'énergie est disponible pour être cédée à l'environnement sous forme de chaleur . Dans le cas de cette réaction , l'énergie libérée sous forme de chaleur est supérieure à la diminution d'énergie interne du système.
- Relation entre Qp et Qv à même température
supposons une réaction entre gaz parfait qui se produit, à une t° donnée, d'une part à pression constante, d'autre part à volume constant. La variation d'énergie interne est la même dans les deux cas ( loi de Joule : l'énergie interne d'un gaz parfait ne dépend que de la t°, donc ?U = 0 à t° constante )
Or , QV = UB - UA
QP = HB -HA = UB - UA + P. (VB -VA)
Il faut donc estimer le terme P. (VB -VA).
soit la réaction :
i J + j J ------> k K + l L
A B
i, j, k, l étant les nombres de moles des composés gazeux I,J,K,L :
P.VA = (i+j)RT
P.VB = (k+l)RT sont vérifiées et par soustraction on a : P . (VB-VA) = ?nRT
?n est la variation du nombre de moles au cours de la transformation d'où on tire : QP = QV + ?nRT
R = 0,082057 l.atm/deg.mole.
= 1,98725 cal/deg.mole.
= 2.10-3 Kcal/deg.mole
Exemple :
Soit la réaction CO + ½ O2 ---> CO2
Connaissant ?H à 298°K et sous Patm : ? H = Qp = - 67,6 Kcal., on peut calculer QV
Dans ce cas : ?n = - 0,5 d'où QV = QP - ?nRT = - 67,6 + 0,5 . 2 . 298 . 10-3 = - 67,3 Kcal
Il est logique que QV soit supérieure à QP car, à P constante, le système reçoit du travail.
La correction du au travail, faible dans ce cas devient plus importante si la t° est élevée ou quand Qp et Qv sont petits.
Pour le calcul de ?n , il faut faire attention aux conditions expérimentales , à l'état physique des substances .
Ainsi, pour H2 + 1/2 O2 -----> H2O sous pression atmosphérique , pour t supérieure à 100°C, ?n = 1 - 1/2 -1 = - 0,5, par contre pour t inférieure à 100°C, l'eau est liquide et ?n = -1/2 - 1 = - 1,5 Par la suite, nous nous intéresseront essentiellement aux chaleurs de réaction à pression constante , en sachant qu'il est facile d'en déduire Qv, à condition d'assimiler les gaz à des gaz parfaits.
- Variation de Qp et de Qv en fonction de la température.
La variation de la température modifie les vibrations inter et intra-moléculaires donc les stabilités des liaisons chimiques d'un composé. Les chaleurs de réaction , résultant de la différence de stabilité entre produits et réactifs seront donc fonction de la température.
On peut également se poser la question : connaissant la variation d'enthalpie d'une réaction pour une température T1, peut-on la calculer pour une température T2 ou doit on effectuer une nouvelle détermination expérimentale ?
Pour traiter ce sujet, nous avons besoin de la notion de capacité calorifique molaire.
- Capacités calorifiques molaires
La capacité calorifique molaire à pression constante Cp est la quantité de chaleur nécessaire pour élever , à pression constante , la t° d'une mole du corps considéré de 1°.
De là, la variation d'enthalpie dH liée à une élévation de t°dT d'un corps à pression constante est : dH = Cp . dT
La capacité calorifique molaire à volume constant Cv est définie de manière analogue, et la variation d'énergie interne dU correspondant à une élévation de t° dT à volume constant est dU = Cv . dT
Ces valeurs reflètent les possibilités de vibration, de translation et de rotation de chaque composé.
Les capacités calorifiques dépendent de la t° de manière trop complexe pour pouvoir être déterminées par calcul . On les exprime par une loi empirique du type :
Cp = a + b.T + c.T2 +...
a,b,c... sont déterminés expérimentalement pour chaque composé dans l'état physique correspondant, T est la t° absolue.
Toutefois, pour les gaz monoatomiques, les capacités calorifiques molaires peuvent être calculées facilement , La seule conséquence d'une élévation de t° est en effet d'augmenter l'énergie de translation des molécules vu qu'il n'y a aucune autre possibilité de rotation ou vibration. Or la théorie cinétique des gaz ( parfaits ) montre que l'énergie cinétique moyenne d'une particule ( molécule ) est égale à : ![]()
où k est la constante de Boltzmann
Pour une môle on aura donc : ![]()
Donc, pouir élever à volume constant la t° d'une môle de gaz parfait monoatomique de ? T, il faut lui fournir une quantité de chaleur : Q = ?T.3/2 R et si ?T = 1°, Q = Cv = R.3/2
Pour calculer Cp, il faut tenir compte du travail échangé, en appliquant la relation : ? H = ? U + P.? V
Dans notre cas particulier ( 1 mole et 1°)
? H = Cp
?U = Cv = R . 3/2
et de PV = nRT on tire P. ? V = R . ?T = R ( car ?T = 1° )
D'où Cp = R.3/2 + R = R.5/2, soit environ 5 calories par moles . Valeur vérifiée pour les gaz parfaits monoatomiques ( gaz rares ) comme le montre le tableau suivant :
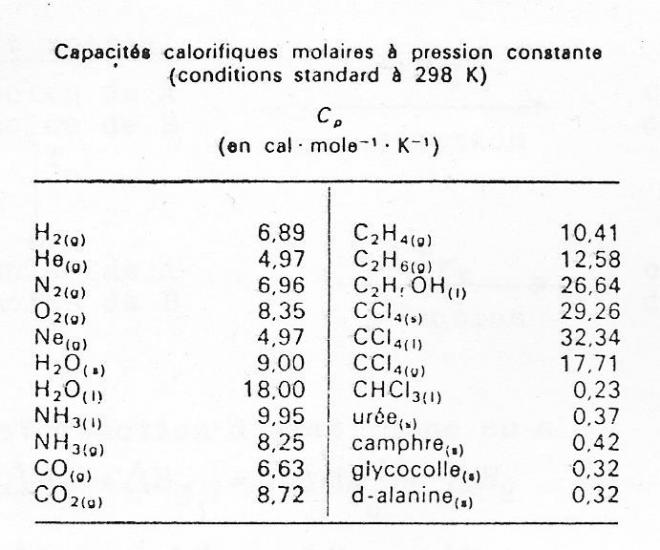
La connaissance des capacités calorifiques molaires à pression constante permet de calculer la variation d'enthalpie accompagnant une élévation de t° , à P constante, pour n moles d'un corps pur passant de T1 à T2 :
![]()
De même, à volume constant, on aurait :
![]()
Remarque : Cp et Cv dépendant de l'état physique de la substance , il ne peut y avoir de changement de phase du composé dans l'intervalle de temps considéré. A partir de là, on pourra calculer les variations d'enthalpie lors d'une réaction chimique en fonction de la température.
- Relation de Kirchoff
Soit une réaction chimique
aA + bB ----> cC + dD
Supposons connue la variation d'enthalpie correspondante pour une t° T0 (?HT) .. On se propose de calculer ?H pour une autre t° T1 (?HT1) . Les composés A,B,C,D sont supposés ne pas changer
Date de dernière mise à jour : 29/11/2018
Ajouter un commentaire