
Chimie générale 11
Cette page comporte deux niveaux de compréhension : La première partie est plus formelle et plus moderne que la seconde, elle est basée sur l'enseignement de chimie générale conféré à partir de l'ouvrage" concentré de chimie" de johan Wouters édité aux presses universitaires de Namur en premier Baccalauréat en sciences chimiques en sciences biologiques et en sciences géologiques ( actuel). Le second niveau correspond à un enseignement de chimie en première candidature d'ingénieur industriel conféré par le Prof. De Cocq à l'époque (1988)
1)INTRODUCTION
Nous tenons compte de deux modèles de l’atome :
- Le modèle de Bohr :
L’électron est une particule et se meut sur des trajectoires concentriques autour du noyau.
Chaque niveau est quantifié énergétiquement : niveaux K L M N O P Q ou n1, n2, n3, n4, n5, n6, n7,
Ce mode d’explication permet par la croix de Lewis de représenter la distribution des électrons sur le niveau énergétique de valence et d’expliquer beaucoup de liaisons.
Plus on s’éloigne du noyau, moins les niveaux ont d’énergie, moins les électrons sont retenus et plus ils ont de potentiel de mouvement. Plus on s’éloigne du noyau plus les électrons ont d’énergie.
La liaison est la mise en commun de deux électrons
- Le modèle de Schrödinger
Modèle strictement mathématique, on ne peut pas vérifier ses résultats expérimentalement.
On caractérise l’électron par une fonction d’onde Ψ que l’on tire de l’équation de Schrödinger, telle que la probabilité de présence s’obtient par intégration sur un petit volume de la densité de probabilité de présence de l’électron. Ψ2 = finalement P = Il n’est pas possible de déterminer un mouvement, mais bien une probabilité de présence dans un volume. C’est ainsi que nous avons étudié la notion d’orbitale atomique.
2) TYPES DE COVALENCES
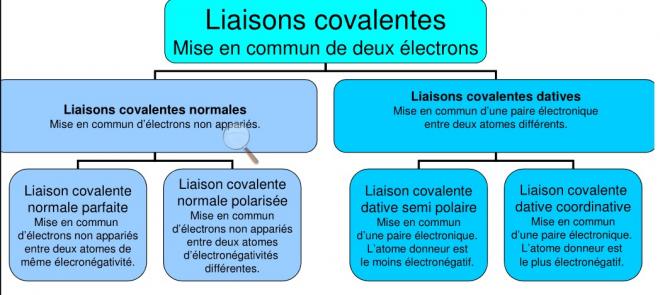
2.1 Covalences normales
Schéma de liaisons covalentes normales
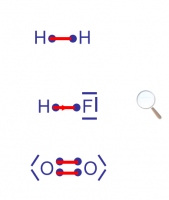
?
les trois types de liaisons covalentes reprises dans cette figure, correspondent successivement à
1) Liaison covalente normale parfaite
2) Liaison covalente normale polarisée
3) Liaison covalente double
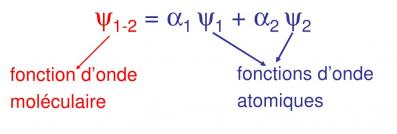
De manière condensée on a : HΨ = E.Ψ et Ψ2 = , équations valables pour les orbitales atomiques et les orbitales moléculaires, pour les atomes et les molécules
|
ATOME |
MOLECULE |
|
Fonction d’onde atomique : Ψ
Niveaux d’énergie atomique : E
Orbitale atomique : Ψ2
|
Fonction d’onde moléculaire : Ψ
Niveaux d’énergie moléculaire : E
orbitale moléculaire : Ψ2
|
On ne peut traiter que le cas de H2 de manière totalement mathématique, pour les autres molécules, les liaisons doivent être traitées avec des approximations.
3. La méthode d’approximation utilisée : Méthode LCAO (Linear Combination of Atomic Orbitals)
On considère que l’orbitale moléculaire d’une molécule à deux atomes est une combinaison linéaire des orbitales atomiques de chaque atome constitutif de la molécule
Cas particulier de la molécule de di-hydrogène

Chaque atome d’H a une orbitale 1s1 on voit que près du noyau de l’atome Ψ1s est maximum = fonction d’onde de l’atome àorbitale de l’atome
Connaissant Ψ1 et Ψ2 qui pour H2 sont les mêmes ainsi que la détermination mathématique de Ψ1-2 on détermine α1 et α2. Selon leurs valeurs on aura les deux possibilités pour l’orbitale moléculaire
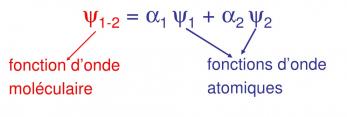
![]() Nous allons maintenant représenter graphiquement l’interpénétration des deux atomes d’H en approchant leurs deux graphiques de la fonction d’onde en fonction de R en abscisse dans le cas ou :
Nous allons maintenant représenter graphiquement l’interpénétration des deux atomes d’H en approchant leurs deux graphiques de la fonction d’onde en fonction de R en abscisse dans le cas ou :
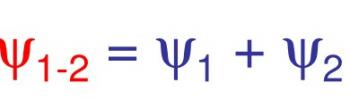
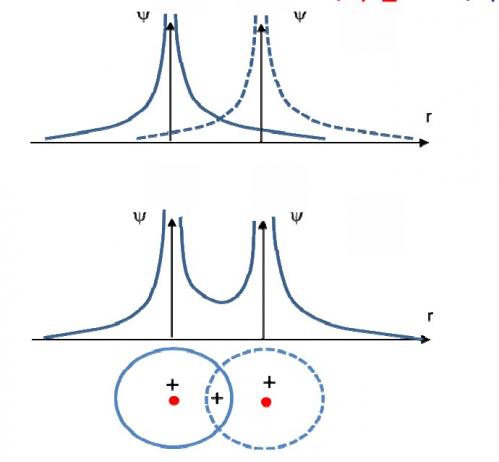
On voit qu’entre les deux atomes, sur la droite qui joint les deux noyaux la fonction d’onde a une valeur positive finie, à l’extérieur elle tend vers l’infini près des atomes et décroît très rapidement vers 0, ce qui ne correspond pas à une liaison
Si on fait pareil dans le cas où :

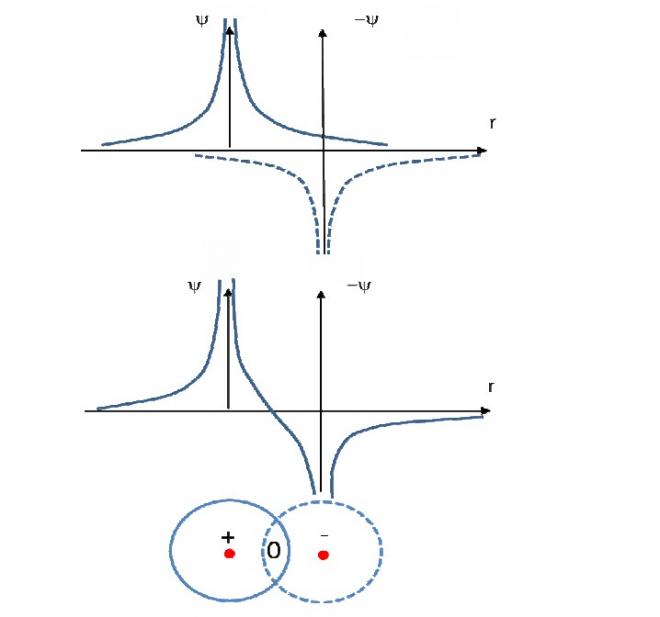
On voit dans ce cas qu’entre les deux atomes, la fonction d’onde s’annule, il n’y a donc pas liaison quoique l’orbitale existe, nous verrons que c’est une orbitale moléculaire non liante. On attribue le signe – aux orbitales dont la fonction d’onde est négative et le signe + lorsque la fonction d’onde est au-dessus de l’axe de R . Si une orbitale + et une orbitale – se « rencontrent » il n’y aura pas liaison.
En portant maintenant les densités de probabilité dans les deux cas sur un graphique en fonction de R on obtient maintenant
dP/dV = ( Ψ1 + Ψ2 )2 àorbitale moléculaire liante et
dP/dV = ( Ψ1 - Ψ2 )2 àorbitale moléculaire non liante, voyons ces cas
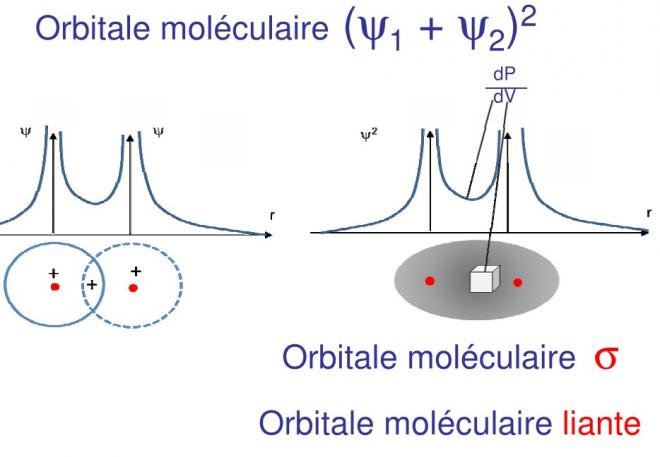
La densité de probabilité résultante est positive et finie entre les deux atomes, il y a une concentration d’électrons entre les deux atomes, c’est l’orbitale moléculaire liante. Lorsque l’on augmente le volume, la probabilité augmente
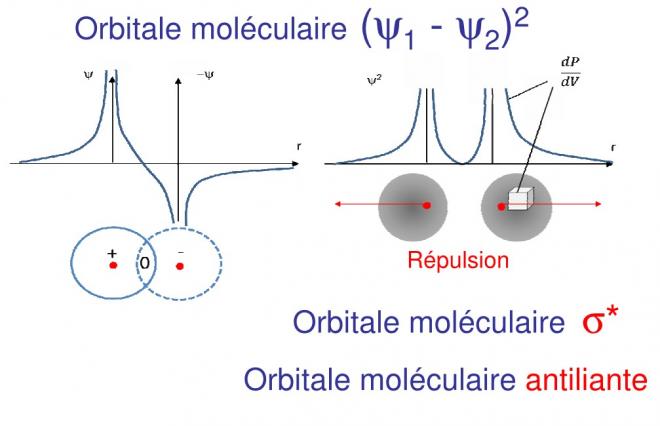
Dans ce second cas, entre les atomes la densité de probabilité s’annule, c’est-à-dire que si l’on augmente le volume, la probabilité n’augmente pas.
Maintenant que nous connaissons la fonction d’onde moléculaire de H2, on peut trouver théoriquement E (énergie) des orbitales moléculaires à partir de HΨ = E.Ψ
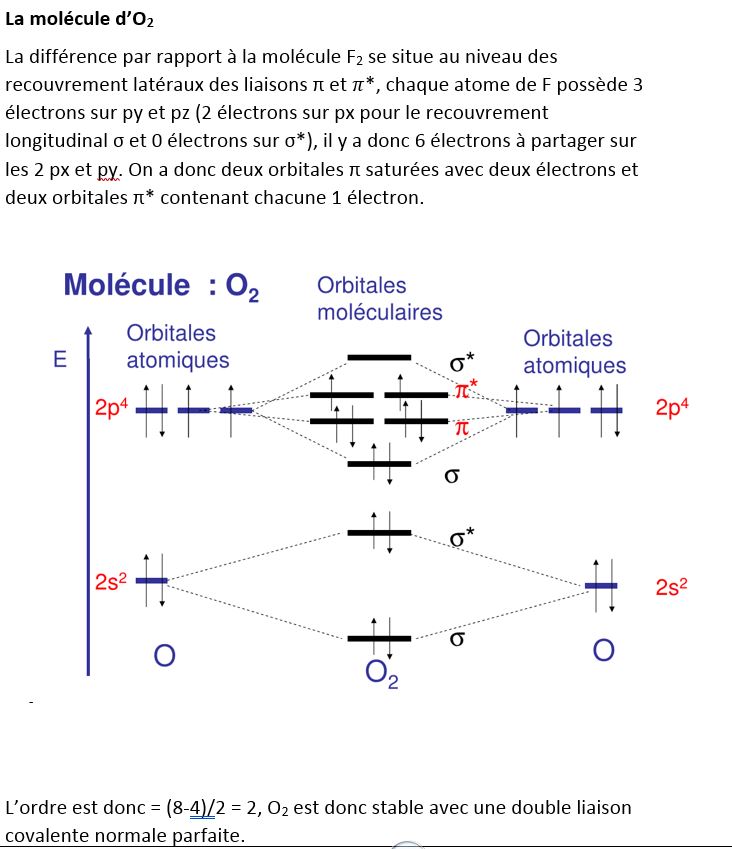
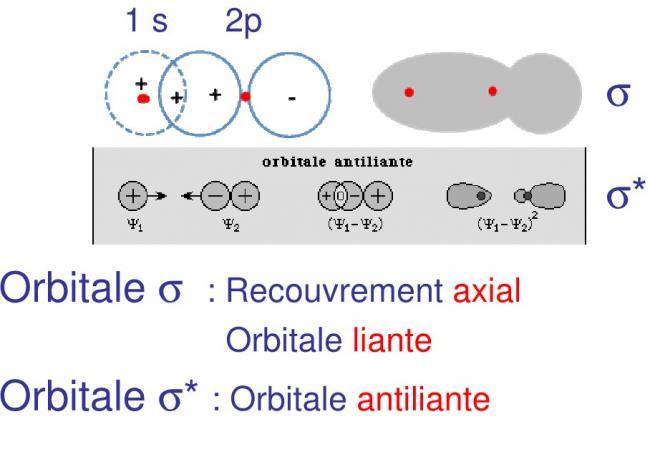
4.Diagramme des niveaux d’énergie moléculaire pour la covalence normale.
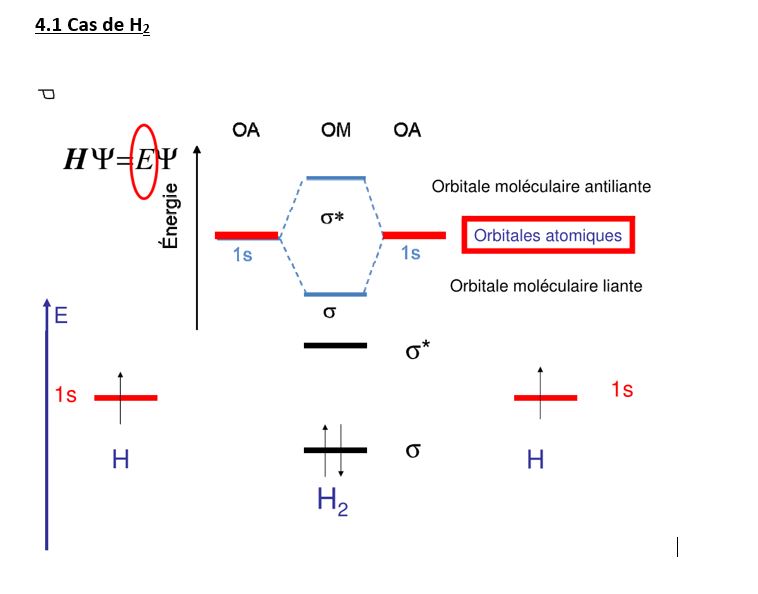
L’orbitale moléculaire comportant une liaison σ (liante) ayant un niveau d’énergie plus faible, se rempli en premier lieu car elle est plus stable. Les électrons de chaque orbitale atomique 1s1 s’apparient pour former cette orbitale liante. L’autre orbitale σ* (non liante) n’a plus d’électrons à sa disposition pour se remplir
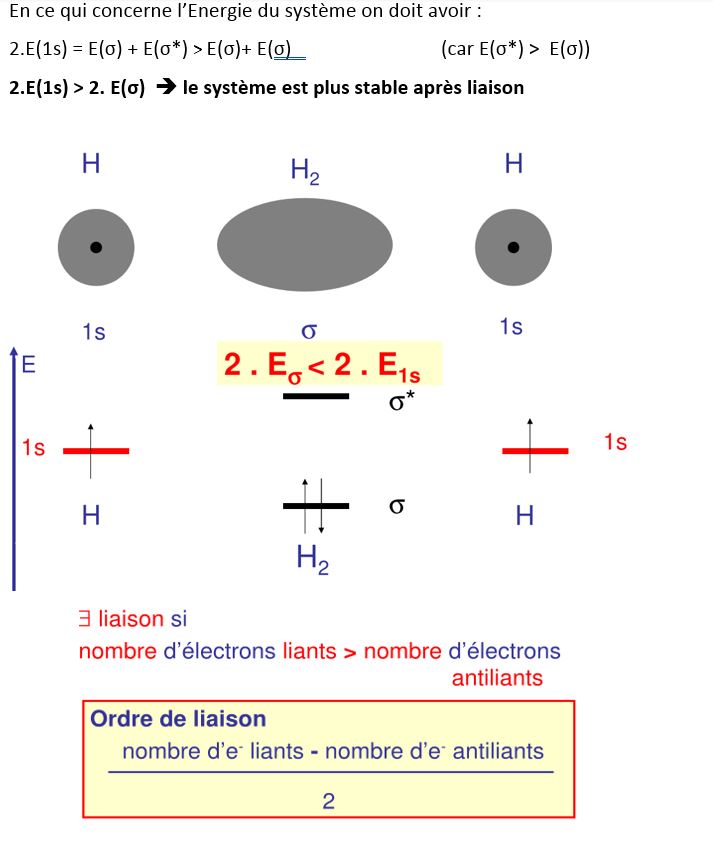
Dans le cas de H2 : on a n (électrons liants) = 2 et n (électrons non liants) = 0
(2-0)/2 = 1 l’ordre de liaison = 1
4.2 Pour la molécule He2
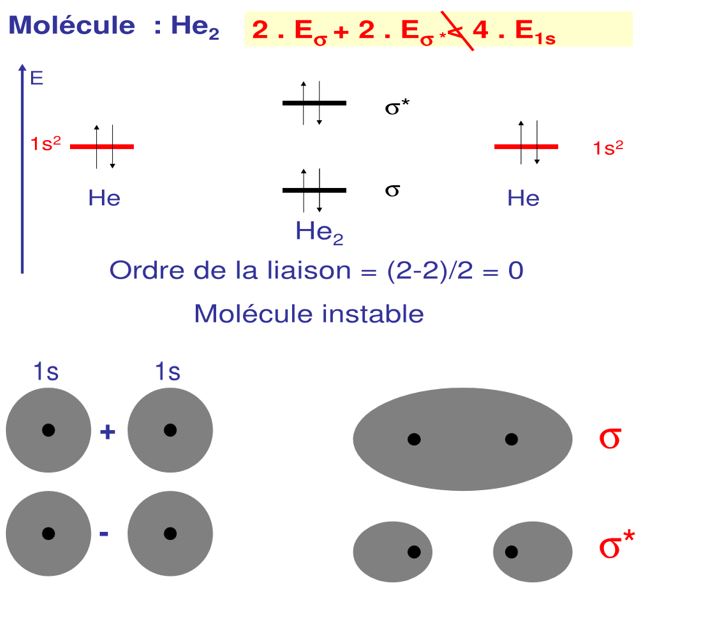
Molécule très instable
Avant liaison on a en tout 4 électrons sur 1s correspondant à 4 électrons d’énergie E1s = 4. E(1s)
Après liaison on a 2 électrons sur σ et 2 électrons sur σ*, en énergie ça donne : 2.E(σ) + 2. E(σ*). On a cependant un système instable donc :
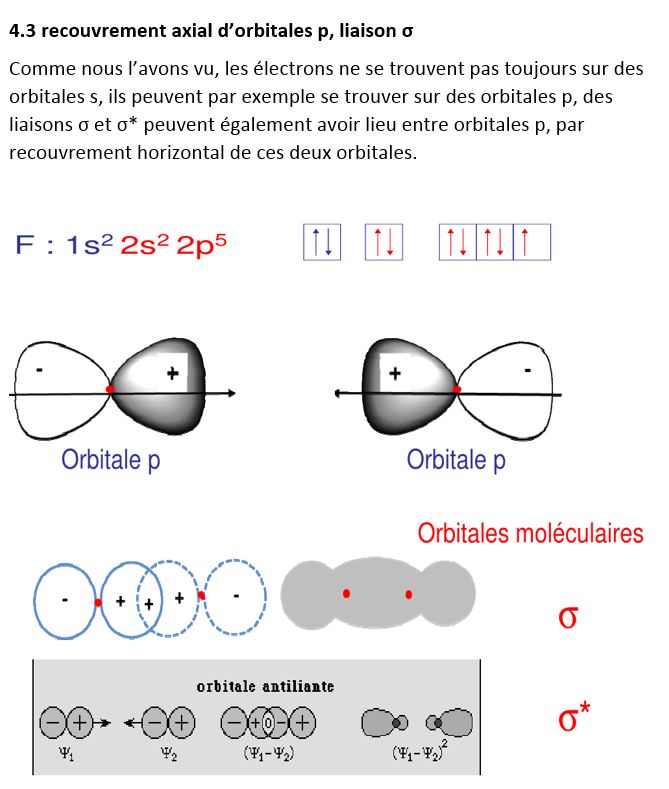
L’application du modèle linéaire donne un résultat différent pour les orbitales p qui comportent un lobe positif et un lobe négatif.
On voit qu’il n’y a que les fonctions d’onde associées de même signe qui font liaison, positif et négatif donnent une fonction d’onde nulle entre les deux atomes, 4 possibilités :
+ - ----- - + et - + ------ + - : OM liantes
+ - ----- + - et - + ------- - + : OM anti-liantes.
Dans les cours théoriques, on ne représente que - + ------ + - comme liante et une des deux possibilités pour les anti-liantes - + ------- - + , ce qui revient au même.
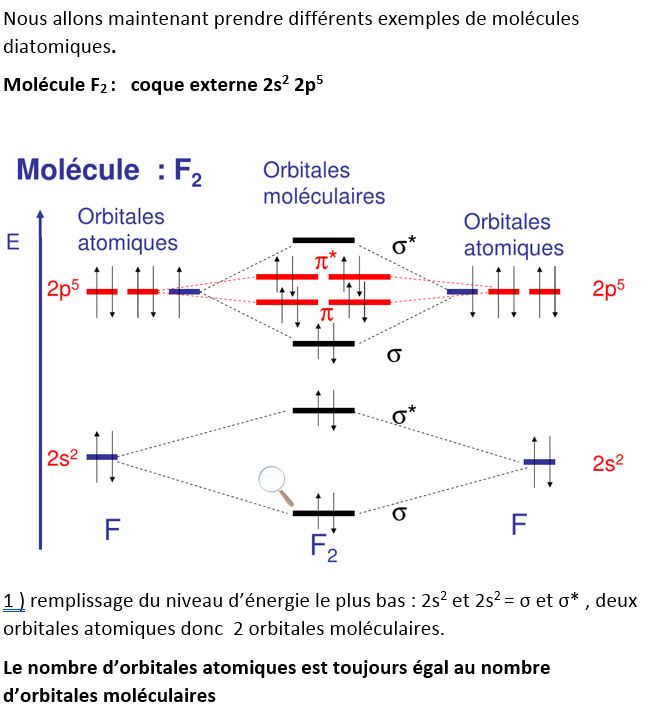
Exemple de F2, on ne représente que les niveaux de la couche externe
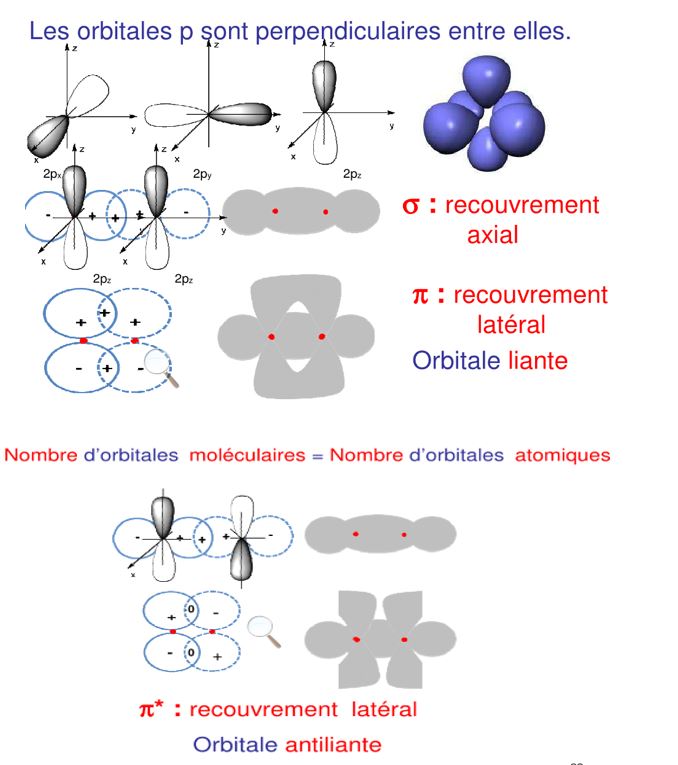
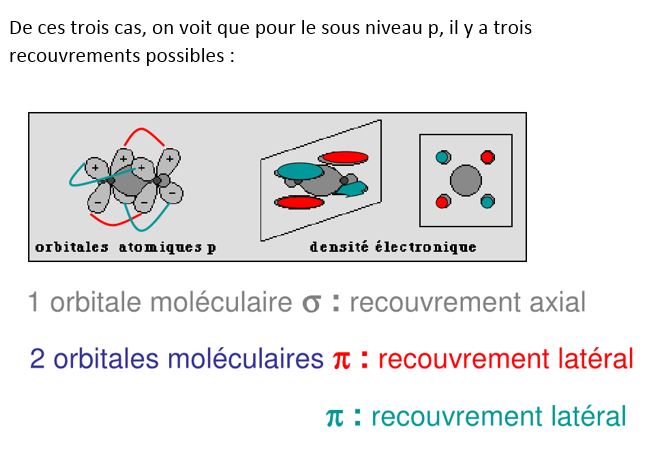
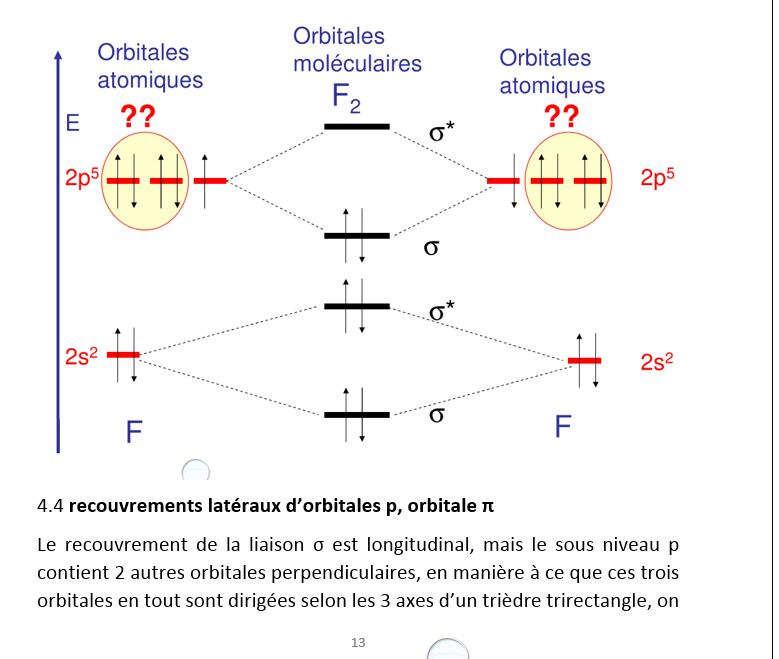 a px, py, pz. il y aura donc 2 possibilités ( en py et pz si px est le recouvrement longitudinal σ ) de recouvrement latéral après le recouvrement longitudinal, ce sont les liaisons π.Les orbitales π peuvent également être liantes et non liantes (π et π*)
a px, py, pz. il y aura donc 2 possibilités ( en py et pz si px est le recouvrement longitudinal σ ) de recouvrement latéral après le recouvrement longitudinal, ce sont les liaisons π.Les orbitales π peuvent également être liantes et non liantes (π et π*)


![]()
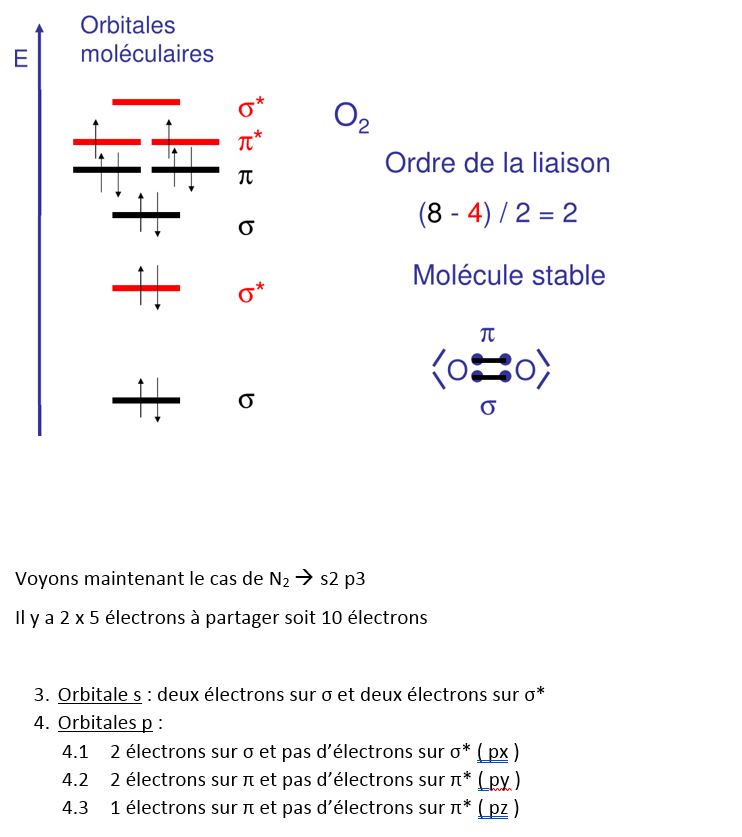

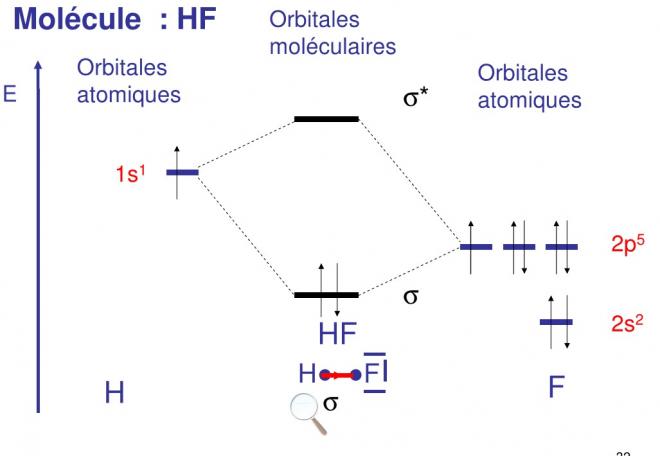
Recouvrement possible. Ordre = (2-0)/2 = 1 une liaison covalente normale polaire , HF est stable.
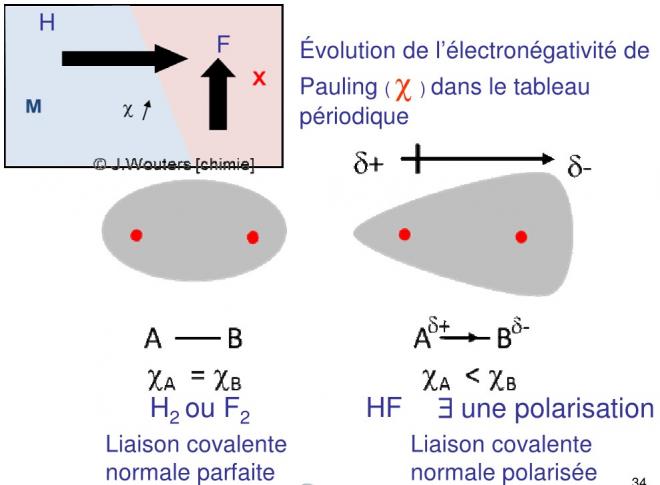
La polarité de la covalence normale polaire est provoquée par la différence d’électronégativité des deux atomes, F est plus électronégatif que h donc le doublet d’électrons sera délocalisé vers F qui aura un résidu de charge δ- et donc H un résidu de charge δ+
La figure suivante compare les covalences normales parfaite et polaire
5.Les liaison covalentes datives (semi-polaires et coordinatives)
5.1Comparaison entre covalence normale et dative
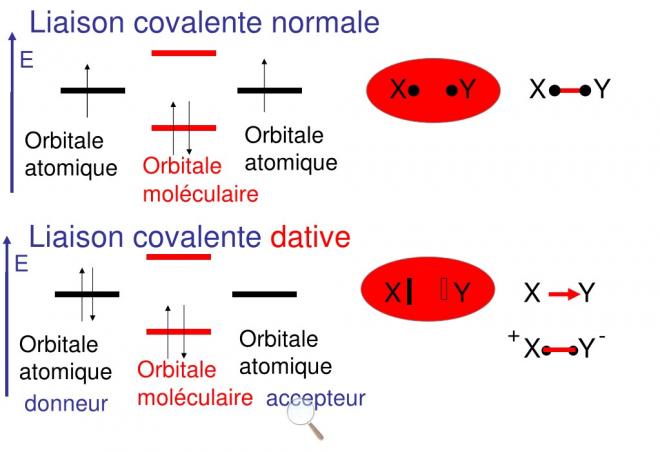 5.2 Les liaisons covalentes datives semi-polaires ou coordinatives se définissent à partir de l’électronégativité du donneur et de l’accepteur
5.2 Les liaisons covalentes datives semi-polaires ou coordinatives se définissent à partir de l’électronégativité du donneur et de l’accepteur

- Liaison covalente dative semi-polaire
Dans une covalence dative, un doublet d’électron est cédé par un donneur à un accepteur qui possède une orbitale vide (case quantique vide)
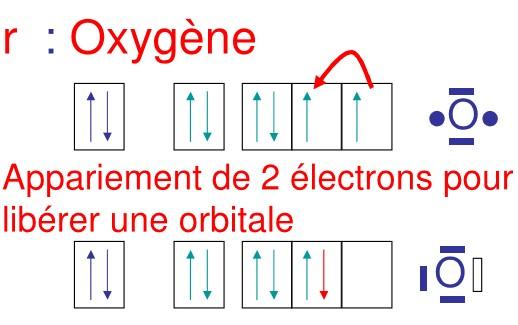
Dans la semi-polaire, l’électronégativité de l’accepteur est supérieure à l’électronégativité du donneur. Souvent pour ménager une orbitale vide ou case quantique vide, il peut y avoir appariement de 2 électrons dans le niveau ( p par exemple ), ce qui donne une case vide
Dans ce cas précis de covalence dative semi-polaire l’accepteur est souvent l’oxygène

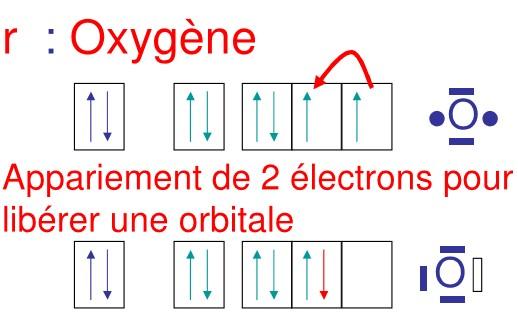
Soit E1 : énergie d’appariement, énergie dépensée par le système donc négative
Soit E2 : énergie libérée lors de la formation de la liaison par le système
E1<E2 à E2 + (-E,) est positive globalement le processus a un bilan positif, E2 étant suffisante pour permettre E1.
 EXEMPLES
EXEMPLES
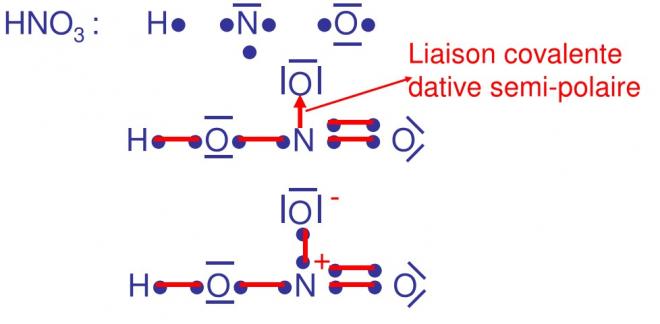
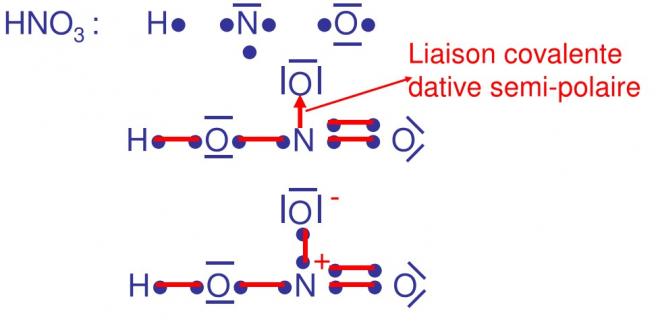
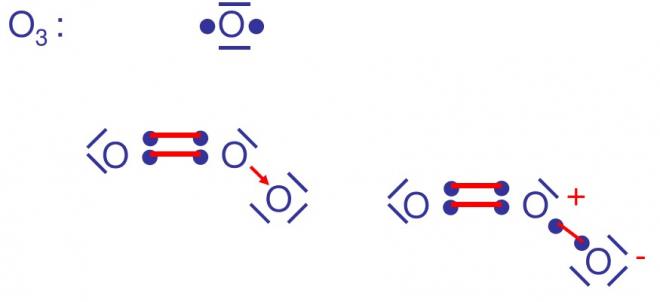
ON ATTRIBUE UNE CHARGE POSITIVE AU DONNEUR ET UNE CHARGE NEGATIVE A L’ACCEPTEUR, le donneur possédait un doublet d’électrons qui finalement se retrouve en commun entre donneur et accepteur. On considère que le doublet est partage comme dans une covalence normale et qu’en fait un électron est au donneur et un électron à l’accepteur ; par rapport à son état avent liaison, le donneur a perdu un électron donc une charge +1 , l’accepteur quant à lui, gagne un électron donc une charge -1

Nous avons vu en haut de la page 26 que la liaison dative semi polaire chargeait la molécule de manière globale, il faut également tenir compte de la polarisation de la liaison.
5.3Liaison covalente dative coordinative
Dans ce cas, l’accepteur est moins électronégatif que le donneur de doublet.
Prenons le cas de NH3 auquel s’ajoute un proton (son orbitale 1s est vide et sa charge est une fois positive) pour donner NH4+. Comme dans le cas des datives semi-polaires un atome gagne un électron et l’autre en perd un (partage du doublet), donc une charge apparaît : positive sur le donneur et négative sur l’accepteur. Dans ce cas, le proton est l’accepteur et est chargé positivement, il va récupérer une charge négative par rapport à son état de proton et sa charge va devenir nulle.
- La notion de nombre d’oxydation
Ce nombre correspond à la valence affectée du signe plus ou moins. Ce signe correspond aux résidus de charges portés par les atomes lors de la liaison. La dative provoque 2 résidus de charge à chaque atome concerné. Le nombre d’oxydation correspond aussi au nombre d’électrons impliqués dans la liaison.
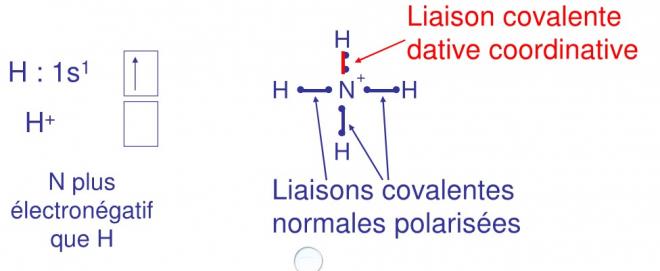 On a un cas similaire pour la formation d’H3o+ à partir de H2O
On a un cas similaire pour la formation d’H3o+ à partir de H2O
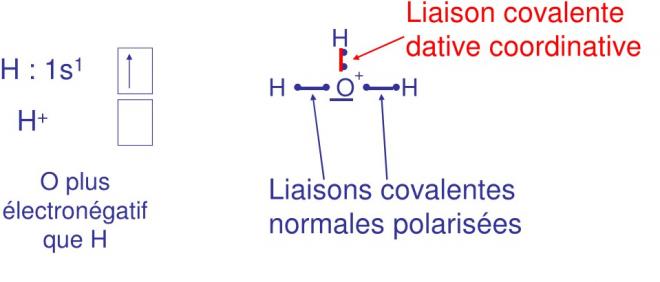 Les nombres d’oxydation ou étages d’oxydation se calculent de la même manière que précédemment, mais une dative coordinative ne change pas ce nombre d’oxydation
Les nombres d’oxydation ou étages d’oxydation se calculent de la même manière que précédemment, mais une dative coordinative ne change pas ce nombre d’oxydation
 On peut tenter à titre d’exercice de construire la structure des complexes métalliques suivants :
On peut tenter à titre d’exercice de construire la structure des complexes métalliques suivants :

- Polarisation des liaisons
- Molécules diatomiques symétriques : apolaires
Dans le cas de molécule diatomiques symétriques comme H2, Cl2, O2 :
- le centre résultant des charges électriques se trouve au milieu de la droite représentant la liaison entre les deux atomes identiques,
- le doublet d’électrons de liaison est attiré avec la même force dans les deux sens puisque les électronégativités sont identiques.
- Les deux nuages électroniques s’interpénètrent pour donner un nuage symétrique par rapport à la droite de liaison et le plan qui lui est perpendiculaire en son milieu. Les centres des charges positives et négatives (barycentre) de chaque nuage vont coïncider au centre de la droite de la liaison. La molécule sera apolaire.
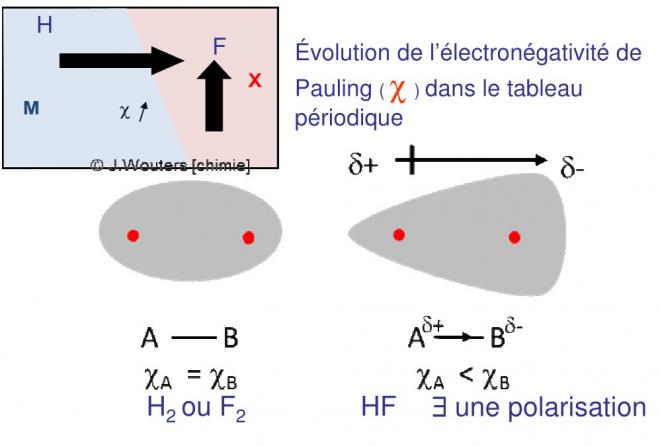
6.2 molécules diatomiques dissymétriques : polaires
6.2.1 caractéristiques
- Si la molécule est dissymétrique (HCl, par exemple), la différence d’électronégativité va créer une polarisation
-L’atome le plus électronégatif attire plus fortement les électrons qui, en moyenne, se rapprochent de lui
- Le nuage électronique n’est pas symétrique par rapport au plan perpendiculaire à la droite de liaison en son milieu
- Des résidus de charges positives et des résidus de charges négatives apparaissent mais leur barycentre ne va plus coïncider et la molécule sera polaire
6.2.2 moment dipolaire
Le moment dipolaire µ est défini conventionnellement comme étant le produit de la charge q en coulomb de chaque atome, multipliée par la distance d entre ces deux atomes en mètres : µ (C.m) = q (C). d(m) ; on prend pour valeur de q, la charge du proton = |charge de l’électron| = 1,602 . 10-19 C
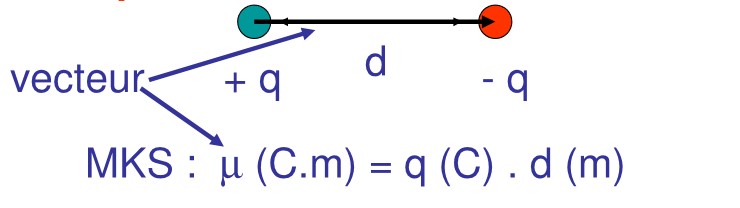 Une unité plus adaptée est de Debye (D) : La distance entre les atomes est de l’ordre de l’Angström Å. On le définit comme suit :
Une unité plus adaptée est de Debye (D) : La distance entre les atomes est de l’ordre de l’Angström Å. On le définit comme suit :

Conventionnellement, on oriente le moment dipolaire du + vers le -
6.2.3 Caractère ionique d’une liaison
Le moment dipolaire réel peut être mesuré expérimentalement = µexp
On définit le pourcentage de caractère ionique de la liaison à partir de la formule :
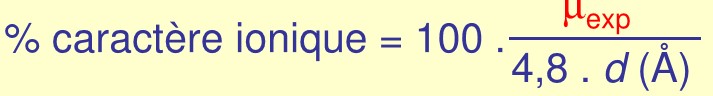 Voici un tableau reprenant les valeurs du pourcentage ionique, des valeurs de d et du moment dipolaire théorique
Voici un tableau reprenant les valeurs du pourcentage ionique, des valeurs de d et du moment dipolaire théorique
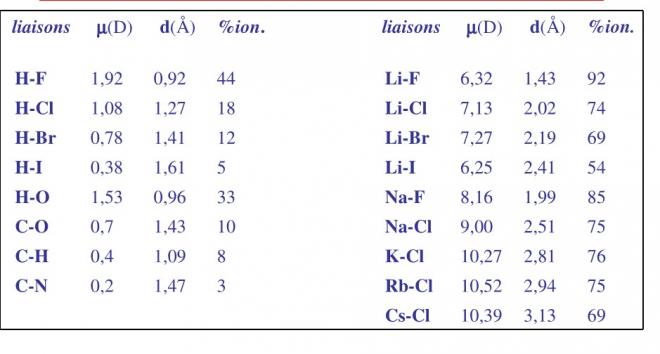
Voyons un exemple de calcul avec la molécule de HCl
Le µthéo correspond à une liaison totalement ionique avec δ = |q électron|
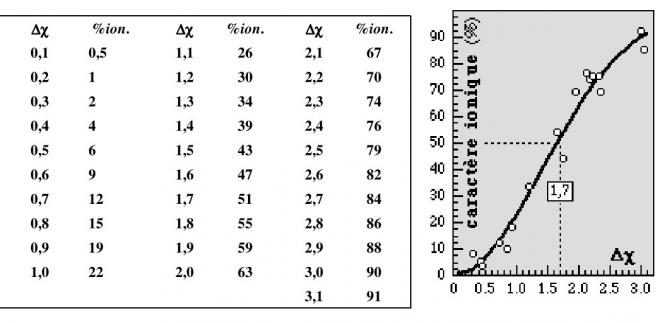
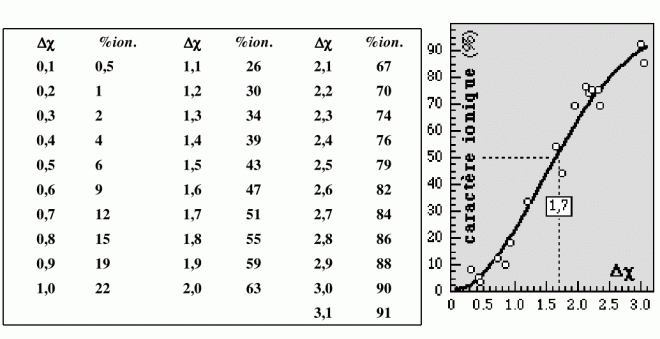
 Le tableau suivant montre l’évolution du %ionique en fonction de Δχ
Le tableau suivant montre l’évolution du %ionique en fonction de Δχ
Rappelons que pour une molécule et pour chaque liaison on a :

La formule doit être adaptée pour des molécules polyatomiques (qui comprennent plusieurs liaisons), µthéo peut être calculé pour chaque liaison, de sorte que µ de la molécule sera égal à la somme vectorielle des µ de chaque liaison
On a :
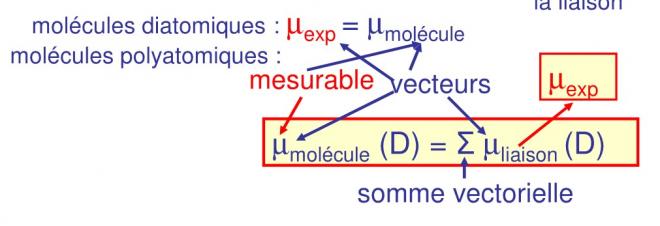
On aura donc le µ théorique de la molécule (µmolécule) à l’aide de cette formule (liaisons 100% ioniques) comme somme vectorielle des µ des liaisons calculés par la formule de Debye. La formule s’applique lorsque l’on utilise le µexp ( que l’on peut mesurer dans des molécules diatomiques polaires) des liaisons ou de la molécule De même pour les différents µ théoriques des liaisons que l’on pourra comparer avec les µ expérimentaux des liaisons et ainsi déterminer le %ionique de chaque liaison. En oubliant pas que µ est un vecteur.
On doit cependant tenir compte de la géométrie de la molécule. Si l’on prend la molécule d’eau, l’angle entre les deux liaisons OH vaut 104,5 °. Le moment total expérimental de la molécule est mesurable, il vaut 187 D. La somme des deux vecteurs µ OH (selon la formule de la page 33) se fait selon la règle du parallélogramme, ça nous donne la direction du µ de la molécule (voir figure), c’est sur cette direction que l’on projettera les µ OH qu’ils soient expérimentaux ou théoriques.
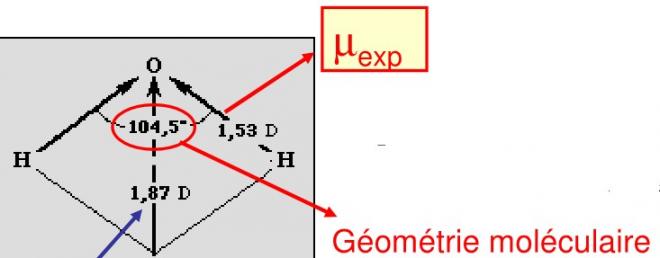
On a :
µOH(D) cos (104,5°/2) + µOH(D) cos (104,5°/2) = 1,87 (D) , ce qui nous donne :
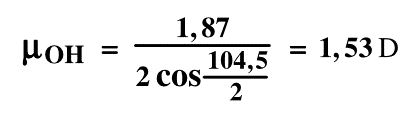
Plusieurs cas peuvent donc se présenter,
Le premier cas correspond à µmolécule (mesurable) ≠ 0, la molécule est polaire avec µ exp ≠ 0 (µ exp = δ . d) et µmolécule = Σ µliaison
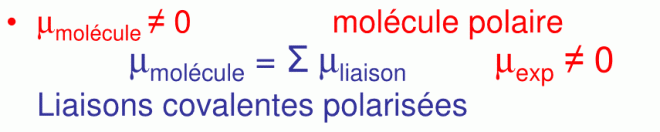
(Datives ou normales)
Toutes les molécules diatomiques de deux atomes différents sont dans ce cas ( HF, HCl…)
Le second cas, c’est lorsque µmolécule (mesurable) = 0, la molécule est non polaire, (µ exp = δ . d) et µmolécule = Σ µliaison
en ce cas il y a deux possibilités :
- Les liaisons ne sont pas polarisées leur moment dipolaire est nul (le moment est nul puisque δ = 0)
µmolécule = 0, µliaison = 0 Σ µliaison = 0,

Le pourcentage ionique est nul : µ exp/(4,8.d) = 0 le µthéorique n’est jamais nul, il correspond à une charge de module 1 des atomes. Donc % ionique = 0/ µthéorique
2) Les liaisons sont polarisées, leur moment dipolaire est ≠ 0 , µmolécule = 0, µliaison ≠ 0, Σ µliaison = 0, mais µexp ≠ 0
 Chaque liaison à son % ionique, dans ce cas on a des liaisons covalentes normales, polarisées, C’est le cas du CO2
Chaque liaison à son % ionique, dans ce cas on a des liaisons covalentes normales, polarisées, C’est le cas du CO2

On peut également prendre l’exemple suivant
 La figure suivante reprend le pourcentage ionique des liaisons en fonction de la différence d’électronégativité - Δχ
La figure suivante reprend le pourcentage ionique des liaisons en fonction de la différence d’électronégativité - Δχ

7.La longueur des liaisons.
7.1 Principe

La détermination de la longueur des liaisons peut se faire directement par des mesures physiques, ces distances internucléaires peuvent cependant être calculées avec une bonne approximation par addition des rayons de covalence des atomes concernés. L’approximation est cependant d’autant moins bonne que la liaison est plus polarisée.
Les liaisons multiples ne permettent cependant pas de calculer la distance internucléaire entre les atomes, la figure suivante montre les distances internucléaires entre quelques liaisons multiples.
7.2 Le rayon covalent.
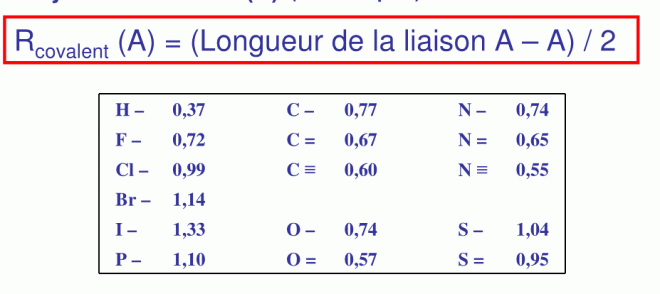
Soit une liaison de covalence entre mêmes atomes, molécule diatomique symétrique A—A
Le rayon de covalence d’un atome est défini comme étant la demi-longueur de cette liaison
 Une petite correction est cependant à apporter, on a :
Une petite correction est cependant à apporter, on a :
 Il faut en fait tenir compte de la différence d’électronégativité des atomes de la molécule Δχ, comme mentionné dans le point 7.1 :
Il faut en fait tenir compte de la différence d’électronégativité des atomes de la molécule Δχ, comme mentionné dans le point 7.1 :
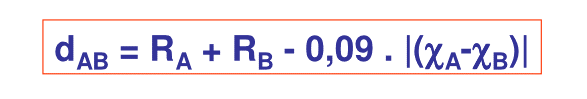
7.3 Évolution du rayon covalent
- Dans une période
 En effet, dans une période, la distance du noyau au niveau des électrons périphérique n’augmente pas avec Z, mais le noyau se charge de plus en plus, ce qui attire les électrons du niveau périphérique
En effet, dans une période, la distance du noyau au niveau des électrons périphérique n’augmente pas avec Z, mais le noyau se charge de plus en plus, ce qui attire les électrons du niveau périphérique
- Dans une famille

En effet, dans une famille, le nombre de niveaux périphériques augmente verticalement vers le bas avec z, le rayon de l’atome augmente avec Z, donc son rayon de covalence également
- Multiplicité des liaisons
Comme nous l’avons mentionné, les liaisons multiples introduisent une discordance avec l’approximation

8.Les liaisons électrovalentes
8.1Limite entre la liaison covalente polarisée et la liaison ionique ( électrovalente)
Nous allons résoudre ce problème de manière conventionnelle

8.2.Caractéristiques physico-chimiques des composés ioniques
8.2.1.Ce sont des composés qui conduisent le courant à l’état fondu
SELS,
BASES HYDROXYLEES
OXYDES BASIQUES
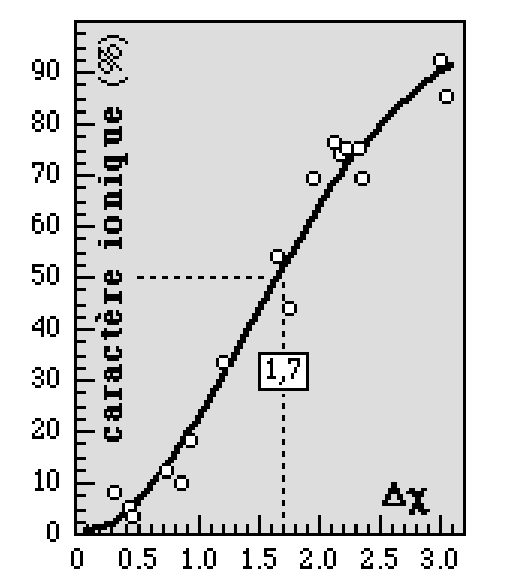
8.2.2 Evolution du caractère ionique d’une liaison en fonction de la différence d’électronégativité entre atomes
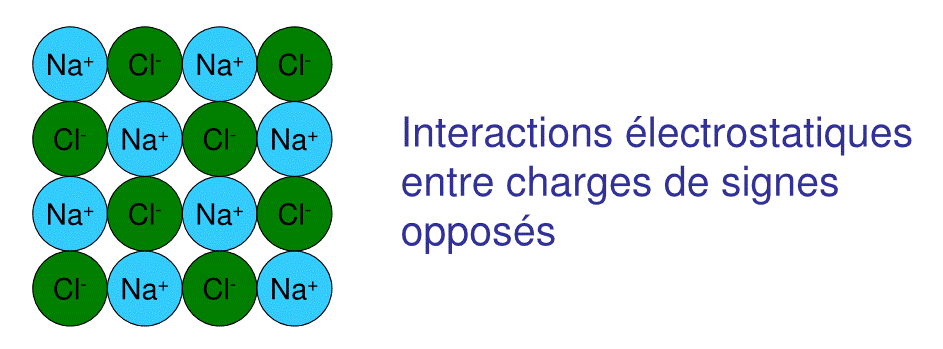
8.2.3.Les liaisons ioniques sont des liaisons non dirigées
A l’opposé des liaison covalentes, les liaisons ioniques ne sont pas dirigées
Les attractions électrostatiques entre charges, se dirige dans toutes les directions, ce qui permet de former un réseau d’atomes comme dans le cas du cristal ionique de NaCl
8.2.4.Pour le NaCl le réseau est tridimensionnel cubique, ici une seule face est représentée.
Voici une meilleure représentation
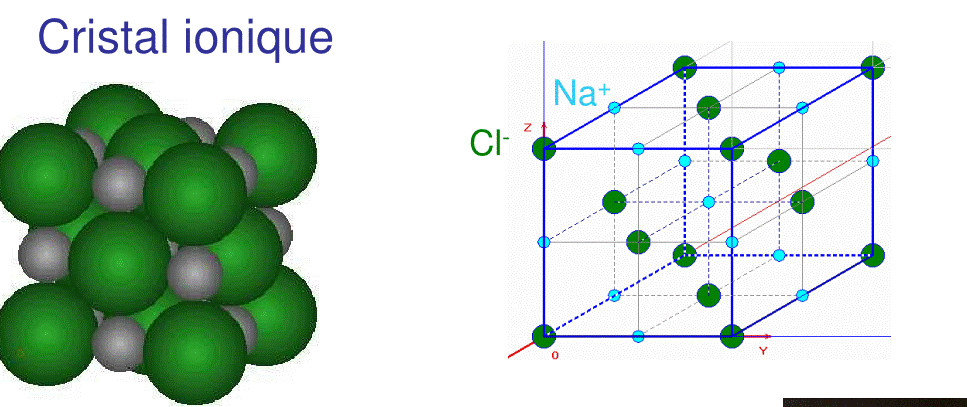
Solides durs et cassants, haute T° de fusion

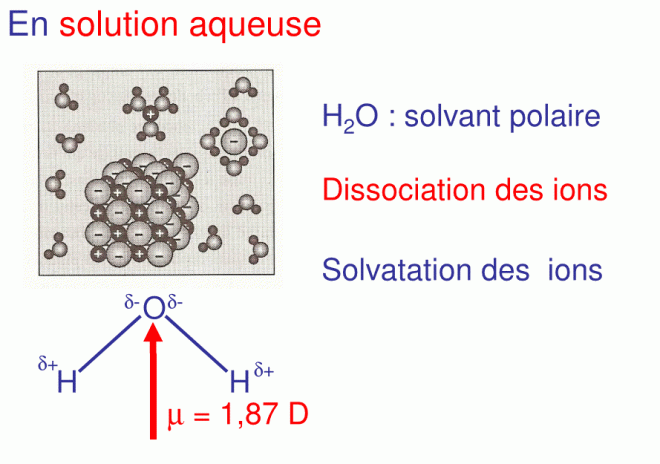
8.2.5.Comportement des composés ioniques en solution aqueuse
En solution aqueuse, les molécules d’eau polaires entrent en interaction électrique avec le cristal de sodium, les ions se dissocient, ainsi les ions Cl- s’entourent de molécules d’eau, ils sont solvatés
8.2.6.Rayons ioniques
Les rayons ioniques sont plus courts ou plus longs entre atomes que dans le cas des covalences.
Dans les liaisons ioniques les atomes sont chargés, ils comportent des charges entières et non des « δ ».
Si c’est un cation, la charge positive de l’atome attire les électrons et le rayon ionique sera plus faible que le rayon covalent, dans le cas d’un anion, les électrons sont repoussés et le rayon ionique est plus grand que le rayon covalent

9.La liaison métallique
Les métaux sont des solides cristallisés.
- Les liaisons entre atomes ne sont pas ioniques car il n’y a pas de différence d’électronégativité entre atomes de même nature
- Des covalences seraient envisageables, sous forme de covalences parfaites non polarisée, mais la relative faiblesse de celles-ci par rapport aux liaisons ioniques ne justifierait pas la dureté des solides métalliques.
Il se pose donc la question de la nature des liaisons métalliques.
Nous disposons d’un modèle pour ces liaisons, voici une version très simplifiée.
Les métaux ont une tendance affirmée à la faible électronégativité, les électrons sont donc peu retenus, on imagine donc qu’ils deviennent mobiles dans le cristal. Les ions positifs constitueront ainsi « l’ossature » du cristal ; les électrons se diffusent dans tout le cristal et « baignent » les ions, leurs mouvements constants sont désordonnés.
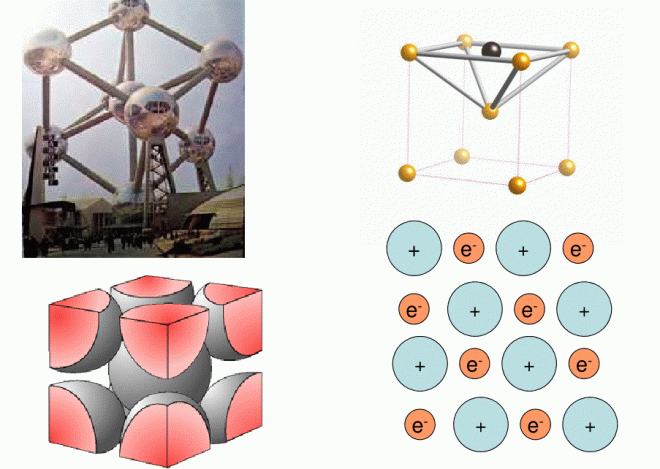
La structure des liaisons métallique justifie :
- La dureté des métaux
- Leur conductibilité électrique.
La liaison atomique ( ou covalente )
1. Généralités - Formation d'une liaison covalente
De nombreuses substance ne se comportent en aucun cas comme des électrolytes , et il ne peut en conséquence être question d'envisager l'interaction ionique pour expliquer leur formation. C'est le cas notamment du méthane ( CH4), du tétrachlorure de carbone ( CCl4) ainsi que des molécules de corps simples.
La stabilité des molécules de ces substances montre que les atomes y sont unis par un type de laison autre que la liaison ionique , mais pourtant solide.
En 1916, pour expliquer la formation de ces molécules, Lewis et langmuir émirent l'hypothèse qu'une telle liaison est due à la mise en commun d'électrons célibataires , de spins opposés , s'associant en doublets, dont chaque partenaire fournit la moitié.
L'exemple le plus simple est celui de la formation de la molécule d'hydrogène par mise en commun de l'électron de chacun des atomes, ce qui peut se représenter :
H - H ou H : H
- ou : étant responsable de la liaison entre les noyaux.
D'autres molécules comme Cl2 , H2O , NH3 , CH4 font également intervenir ce type de liaison, on les représentera en faisant intervenir le tiret _ comme symbole d'un doublet électronique , de la façon suivante :
fonctio
Une fois la liaison formée, si on attribue à chaque atome considéré isolément tous les doublets qui l'entourent, on remarque que la couche électronique externe de chacun des atomes ci dessus a la configuration d'un gaz rare , ce qui doit correspondre à un accroissement de stabilité par rappoprt aux constituants de départ.
En se basant sur ces constatations , Lewis avait énoncé la règle de l'octet mais nous allons voir qu'une fois de plus elle doit être appliquée avec souplesse.
Ainsi le bore ( Z = 5 ) dans BF3 ne présente que 6 électrons sur sa couche externe ( exception par défaut ) .
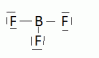
Enfin, dans le cadre de cette théorie de Lewis, il est parfaitement logique que deux atomes puissent échanger plus d'un doublet, avec formation de ce que nous appellerons des liaisons doubles ou triples.
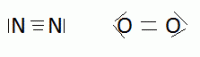 C'est à ce type de liaison qu'a été donné le nom de liaison covalente
C'est à ce type de liaison qu'a été donné le nom de liaison covalente
Dans cette optique, la valence covalente d'un élément sera égale au nombre d'électrons célibataires qu'il peut engager dans des doublets.
Il est un type particulier de liaison covalente qui ne se distingue de la précédente que par le mode de formation. Il s'agit de la covalence coordinative ou semi-polaire. Dans ce cas, la mise en commun du doublet est un acte unilatéral, le doublet étant fourni par un seul des partenaires, mais, une fois formée, nous admettons que la liaison semi-polaire est identique à la liaison covalente .
On la représente par un trait fléché -----> au lieu d'un trait simple , allant du donneur de doublet ( A ) vers l'accepteur (B) :
A ------>B
Mais cette mise en commun confère à l'atome A une charge positive par rapport à son état fondamental et à l'atome b une charge négative. En effet, tout se passe comme si A cédait une partie de son doublet électronique à B . Ceci permet une notation :
A(+) - B(-) ce qui justifie l'appellation de liaison " semi- polaire"
Exemples :
a) un cas typique illustrant la formation des liaisons covalentes normales et semi-polaires est celui de la distribution des liaisons dans les composés organiques nitrés :
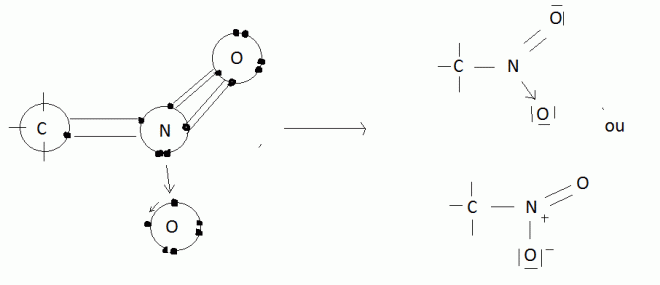 b) Formation de la molécule de SO2
b) Formation de la molécule de SO2
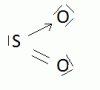 c) Formation de complexes ( où composés de coordination, d'où le nom de valence coordinative ).
c) Formation de complexes ( où composés de coordination, d'où le nom de valence coordinative ).
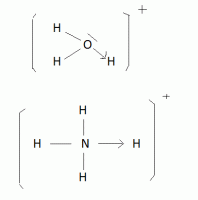 2. Interprétation par les théories de la mécanique ondulatoire
2. Interprétation par les théories de la mécanique ondulatoire
En 1927, soit une dizaine d'années après les travaux de Lewis, Heitler et London analysèrent à la lumière des théories de la mécanique ondulatoire le problème de la formation de la molécule de H2
Les principes dont ils se servirent sont en fait les principes généraux de formation des liaisons atomiques.
L'étude théorique de la formation de cette molécule est faite dans le cadre des cours de physique théorique et mathématique.
Signalons qu'elle est basée sur les principes suivants :
- La distribution des électrons dans une molécule doit obéir à l'équation de Schrödinger, laquelle n'a de solution que pour une suite discontinue de valeurs de l'énergie totale du système , valeurs qui définissent autant de niveaux d'énergie dans une molécule. Les solutions de l'équation de schrödinger pour ces niveaux définissent des zones de probabilité de présence des électrons autour des noyaux de la molécule, zones qui constituent ce que l'on appelle l'orbitale moléculaire .
- La molécule ne peut exister en tant qu'entité stable que si sa formation correspond à une diminution d'énergie par rapport à la somme des énergies des constituants isolés. L'état stable de la molécule correspond au minimum de la courbe d'énergie en fonction de la distance entre atomes constituants.
- Dans chaque orbitale moléculaire , les électrons devront obéir au principe d'exclusion de Pauli et devront donc être antisymétriques.
3. Interprétation des liaisons dans les molécules polyatomiques.
Les interprétations que nous pouvons donner des liaisons interatomiques de covalence et de coordination devront rendre compte de faits établis expérimentalement , à savoir les orientations des différentes liaisons atomiques d'une molécule les unes par rapport aux autres et les valeurs des énergies de liaisons. Pour ce faire, nous nous baserons sur deux grands principes généraux :
a) le principe des liaisons localisées
b) le principe de l'hybridation des orbitales.
4. principe des liaisons localisées : Construction des orbitales moléculaires
a) Reprenons l'exemple le plus simple : celui de la molécule d'hydrogène.
Lors de la formation de la molécule, nous sentons intuitivement que la zone de probabilité de présence du doublet commun sera plus grande entre les noyaux qu'à l'extérieur de ceux - ci .
La plus grande partie de l'énergie de liaison sera fournie par l'échange des deux électrons mis en commun, chacun étant soumis à une force attractive par les deux noyaux simultanément ( voir résonnance ).
Lors de la généralisation de l'étude des liaisons , nous admettrons :
- qu'une liaison atomique concerne seulement deux atomes .
- qu'elle est de même nature que la liaison H-H dans la molécule H2.
- qu'elle est déterminée par des électrons de spins opposés.
- Dans le cas de la molécule d'hydrogène chaque orbitale atomique est caractérisée par une fonction d'onde F exprimant la probabilité de présence de l'électron.
 fig 1 ---------------->
fig 1 ---------------->
Dans l'état fondamental, cette orbitale est une sphère centrée sur le noyau. La formatioin de la molécule s'explique par l'idée de recouvrement ; d'interpénétration des orbitales atomiques avec formation d'une orbitale moléculaire , zone de probabilité de présence des deux électrons assurant la liaison.
Dans le cas de l'hydrogène , l'orbitale moléculaire est intuitivement un pseudo ellipsoïde de révolution autour de l'axe des noyaux . Elle résulte d'une combinaison linéaire des orbitales atomiques . On pourra ainsi obtenir deux niveaux orbitaux selon que les spins des électrons seront antiparallèles ou parallèles. Dans le premier cas , on aura une orbitale moléculaire "liante" avec une forte densité de probabilité des charges entre les noyaux ; dans le second cas, l'orbitale sera dite "anti-liante" avec un espace libre de probabilité de charge nulle entre les noyaux ( voir figure 1 )
b) Structure des molécules diatomiques covalentes homonucléaires
Dans ce cas, les atomes constituants sont les mêmes. lorsque nous rapprochons, comme dans le cas de l'hydrogène deux orbitales atomiques du type s , l'interpénétration se fait suivant l'axe qui joint les noyaux ; l'orbitale moléculaire obtenue , de forme pseudo-sphérique , pouvant être liante ou anti-liante.
A cette orbitale , dont la forme rappelle celle des orbitales tu type s, on a donné le nom de liaison sigme Sigma , l'orbitale antiliante étant caractérisée dans la liaison par une astérisque .
Lors de la formation d'orbitales moléculaires à partir d'orbitales atomiques du type p, il faut tenir compte du fait que ces orbitales atomiques, notées Px, Py et Pz représentent trois zones orthogonales.
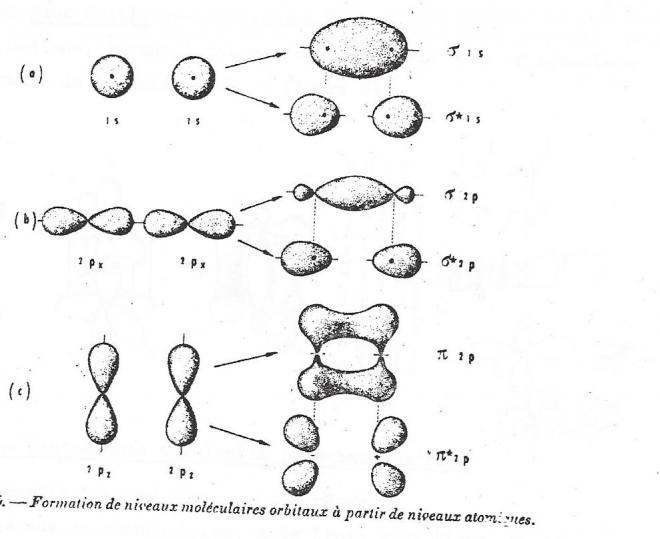 fig 2
fig 2
L'orbitale moléculaire la plus stable que l'on pourra obtenir à partir de ces niveaux est celle correspondant à l'interpénétration maximum suivant la droite joignant les noyaux . L'orbitale moléculaire obtenue a la même symétrie par rapport à la droite joignant les noyaux . L'orbitale moléculaire obtrenue a la même symétrie par rapport à la droite joignant les noyaux que l'orbitale formée à partir de niveaux atomiques s, on la nomme aussi liaison Sigma. Si elle provient de la fusion d'orbitales 2p on la notera Sigma 2p , avec ou sans astérique selon qu'elle est antiliante ou liante. Mais lors de l'élaboration d'une telle liaison, les noyaux se rapprochent.
Pour autant qu'elles ne contiennent qu'un électron, les orbitales Py et Pz peuvent se réunir pour former deux espèces de dirigeables , l'un au dessus, l'autre en dessous de la droite qui joint les noyaux. Cette liaison obtenue par recouvrement latéral des orbitales p a reçu le nom de liaison pi . Elle peut aussi être liante ou antiliante.
Le recouvrement est limité par la forme des orbitales p et ce second type de liaison sera forcément plus faible que la liaison Sigma.
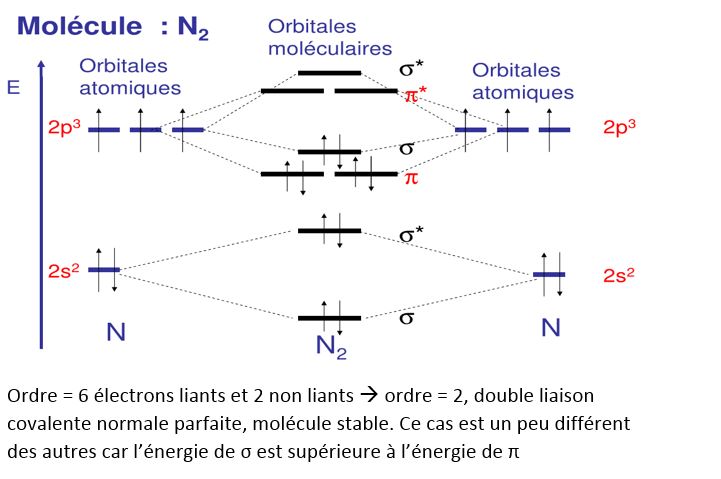
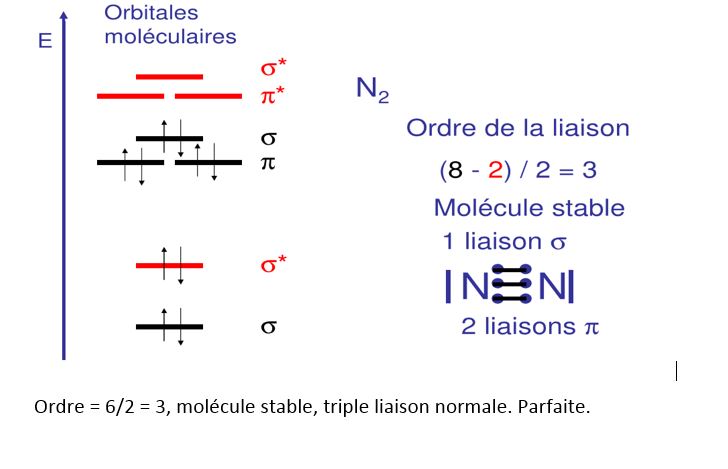
A partir de ces notions , voyons à titre d'exemple quels sont les mécanismes de formation et la structure de la molécule N2 : figure 3.
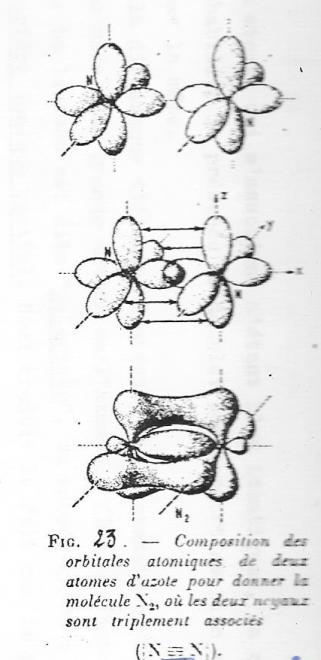 fig 3
fig 3
c) Structure des molécules diatomiques covalentes hétéronucléaires.
Ici les atomes constituants ne sont pas les mêmes.
Les principes généraux ne changent pas, mais trois conditions , pratiquement intuitives doivent être satisfaits pour que les orbitales atomiques puissent se combiner :
- Elles doivent avoir des énergies du même ordre de grandeurs.
- Leur recouvrement doit être aussi grand que possible .
- Elles doivent avoir même symétrie par rapport à l'axe qui les lie .
Considérons un exemple simple, celui de la molécule de HF.
Les structures électroniques des atomes de départ sont respectivement :
H : 1s1 F : 1s2 2s2 2p5
l'orbitale moléculaire représentative de la liaison H-F doit être une combinaison linéaire de l'orbitale atomique 1s de l'hydrogène avec une orbitale atomique du fluor.
Les O.A. 1s2 et 2s2 de celui-ci ont des énergies trop faibles pour le faire et sont d'ailleurs saturées ; c'est l'orbitale 2p ( x,y ou z ) de F qui convient. La liaison sera une liaison Sigma ( fig 4).
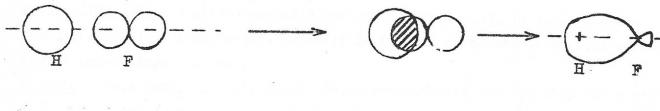 fig 4
fig 4
Les atomes qui s'associent n'étant plus identiques, la répartition de la zone de probabilité de présence du doublet par rapport au plan médian du segment H-F n'est pas le même.
Vu la différence d'étectronégativité des partenaires, la concentration électronique sera plus grande autour de l'atome de fluor qu'autour de l'atome d'hydrogène. La liaison est "polarisée", ou encore qu'on a une liaison covalente à caractère ionique partiel. Ceci traduit le fait que le passage de la liaison du type ionique au type covalent ne se fait pas brusquement mais graduellement.
Ainsi dans le chlorure d'hydrogène , nous écrirons la formule moléculaire HCl en représentant une liaison covalente entre H et Cl.
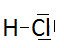 mais nous allons montrer que cette covalence est teintée d'un caractère ionique à raison de 20 %.
mais nous allons montrer que cette covalence est teintée d'un caractère ionique à raison de 20 %.
Bien entendu, ce caractère ionique partiel sera fonction de la différence d'électronégativité entre les partenaires.
On pourra calculer le pourcentage de caractère ionique d'une liaison covalente en utilisant l'échelle des électronégativité selon Pauling de la façon suivante :
- Les éléments les plus éloignés l'un de l'autre au point de vue électronégativité sont le fluor et le césium. La liaison la plus ionique que l'on puisse rencontrer sera celle du fluorure de césium CsF, avec une différence d'électronégativité 4,0 - 0,7 = 3,3.
On la dira de caractère ionique égal à 100 %
Dans ce cas , une différence d'électronégativité entre deux éléments est égale à 1,65, correspondra à 50 % de caractère ionique
- Si la différence d'électronégativité entre deux éléments est supérieure à 1,65 % est la liaison est à caractère ionique prédominant, on écrit la formule sous forme ionique ( Li+Cl- ) .
- Si la différence d'électronégativité est inférieure à 1,65, la liaison est à caractère covalent prédominant, on écrit la formule covalente et on peut calculer le pourcentage ionique :
Exemple : pour BeCl2 , la différence d'électronégativité est 1,5 d'où 100 x 1,5/3,3 = 45 % de participation ionique.
Signalons toutefois que l'utilité primordiale des nombres d'électronégativité est de permettre une comparaison des éléments entre eux , et que les valeurs ne prétendent pas à une exactitude rigoureuse.
d) Orientation des liaisons , caractère dirigé de la covalence.
Considérons maintenant, en nous basant sur les conceptions précédentes la structure des molécules polyatomiques covalentes
1° La molécule d'eau
La structure électronique de l'oxygène (Z = 8 ) : 2s2 2s2 2p4
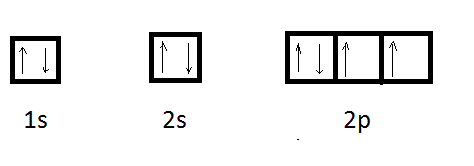 montre bien que sa covalence sera égale à 2.
montre bien que sa covalence sera égale à 2.
On peut donc admettre la formation de la molécule par recouvrement des orbitales 2p non saturées par l'orbitale d'atome d'hydrogène. Ceci va nous conduire à deux orbitales moléculaires dont les axes forment un angle de 90 °.
En pratique, l'expérience montre que l'angle des deux liaisons est de 104° 30 ' , ce qui s'explique parfaitement par la répulsion mutuelle des charges résiduelles portées par les deux atomes d'hydrogène après que les liaisons aient été établies .
De même l'étude physique de H2S et H2Se conduit aux mêmes conclusions mais avec des angles de 92 ° et 90 °, les deux atomes d'hydrogène étant plus éloignés l'un de l'autre, vu le volume croissant des partenaires, et la participation ionique de la liaison étant moindre.
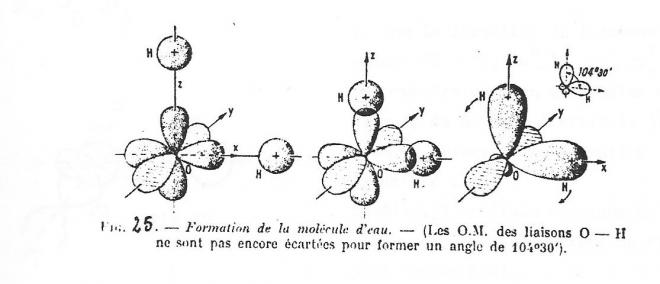 fig 5
fig 5
2° La molécule d'ammoniac
La structure de l'azote ( Z = 7 ) étant : 1s2 2s2 2p3
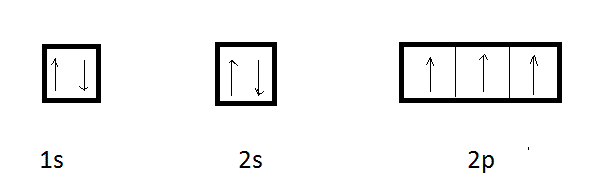 La molécule de NH3 se formerait donc par simple recouvrement des orbitales p incomplètes , orthogonales 2 à 2 par l'orbitale des atomes d'hydrogène. Ici, l'angle entre liaisons sera de 106°45' suite à la répulsion des charges résiduelles des atomes d'hydrogène ( pour PH3 et AsH3 les angles valent respectivement 94 ° et 91°30' ) .
La molécule de NH3 se formerait donc par simple recouvrement des orbitales p incomplètes , orthogonales 2 à 2 par l'orbitale des atomes d'hydrogène. Ici, l'angle entre liaisons sera de 106°45' suite à la répulsion des charges résiduelles des atomes d'hydrogène ( pour PH3 et AsH3 les angles valent respectivement 94 ° et 91°30' ) .
5. Principe d'hybridation des orbitales
L'expression de la structure de certaines molécules par le principe des liaisons localisées va rencontrer quelques difficultés
Pour les résoudre, nous devrons faire appel au principe d'hybridation des orbitales imaginé par Pauling.
a) Ainsi, si nous considérons l'atome de béryllium dans son état fondamental ( Z = 4 ) : 1s2 2s2, il a tous ses électrons appariés et ne devrait donc manifester aucune réactivité .
Toutefois, si on lui fournit une énergie suffisante, l'un des électrons de la couche 2s peut être activé, conduisant à un atome de Be excité, de structure Be* 1s2 2s1 2p1 , qui possède deux orbitales atomiques avec des électrons non appariés et peut former deux liaisons de covalence.
Voyons le formation de liaisons avec le chlore ( Z = 17 , 3s2 3p2x 3p2y 3p1z ) . La première se fera par la fusion de l'orbitale 2p du Be avec l'orbitale 2pz d'un premier chlore, avec formation d'une liaison Sigma.Ensuite, l'orbitale atomique 2s de Be peut encore fixer un atome de chlore pour donner BeCl2
Cette seconde liaison sigma, provenant de la fusion d'une orbitale atomique 2s pourra prendre n'importe quelle orientation autour du noyau de bérryllium, donc par rapport à la première liaison Sigma . De plus la longueur et l'énergie de cette seconde liaison différer de celles de la première.
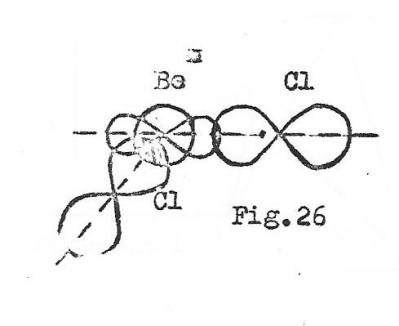 fig 6
fig 6
Or l'étude de BeCl2 aux rayons x montre que la molécule de BeCl2 est linéaire et que les deux chlores y jouent exactement le même rôle , les deux liaisons ont même énergie .
Si la linéarité de la molécule peut encoçre s'interpréter par la répulsion des charges résiduelles des deux chlores, l'aspect énergétique ne trouve qu'une seule expolication : il faut admettre que les orbitales atomiques utilisables du béryllium ne sont plus les orbitales 2s et 2p mais deux orbitales identiques entre elles et différentes des précédentes.
Il y a en fait hybridation des orbitales , c'est à dire fusion des orbitales atomiques avec génération de deux orbitales hybrides, absolument équivalentes , que l'on désigne sp1 ( voir carbone )
Cette notaion prouve que l'hybridation se fait à partir d'une orbitale s et d'une orbitale p . Le nombre d'orbitales hybrides est égal au nombre d'orbitales de départ.
La thermodynamique , sous sa forme du principe de l'état initial et de l'état final , ne s'oppose pas à cette théorie. En effet, les énergies mises en jeu pour passer de l'état initial 1 ( fig 5 ) à l'état final 2 sont indépendantes des transformations subies par le système pour passer d'un état à l'autre.
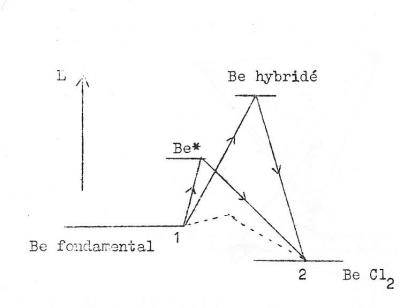 fig 7
fig 7
b) cas du carbone .
L'étude des liaisons du carbone ne peut se justifier qu'à condition de faire appel au phénomène d'hybridation . Elle nous permettra de montrer qu'il y a d'autres types d'hybridation que celui que nous venons d'examiner .
Dans son état fondamental , l'atome de carbone ( Z=6 ) 1s2 2s2 2p2
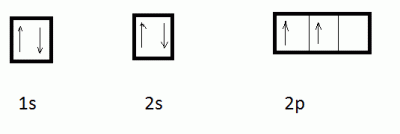 n'a que deux électrons célibataires, il devrait se comporter comme bivalent. Or, toute la chimie organique repose sur sa tétravalence . Par apport d'énergie on peut l'amener dans l'état excité C* 1s2 2s1 2px1 2py1 2pz1 : qui peut expliquer son caractère tétravalent , mais 3 des liaisons serainet orthogonales deux à deux, la quatrième, provenant de l'orbitale atomique 2s ayant une direction quelconque et une énergie différente des 3 autres.
n'a que deux électrons célibataires, il devrait se comporter comme bivalent. Or, toute la chimie organique repose sur sa tétravalence . Par apport d'énergie on peut l'amener dans l'état excité C* 1s2 2s1 2px1 2py1 2pz1 : qui peut expliquer son caractère tétravalent , mais 3 des liaisons serainet orthogonales deux à deux, la quatrième, provenant de l'orbitale atomique 2s ayant une direction quelconque et une énergie différente des 3 autres.
Diverses possibilités vont nous permettre d'expliquer le comportement du carbone
1° Hybridation sp3 - Lorsque le carbone contracte des liaisons avec 4 atomes, il y a fusion de l'orbitale s et de 3 orbitales p pour donner 4 orbitales hybrides équivalentes, notées sp3 qui s'orientent dans l'espace avec un maximum de symétrie c'est à dire vers les somets d'un tétraèdre régulier dont l'atome de carbone occupe le centre, symétrie qui, selon la mécanique ondulatoire correspond au maximum d'énergie des liaisons.
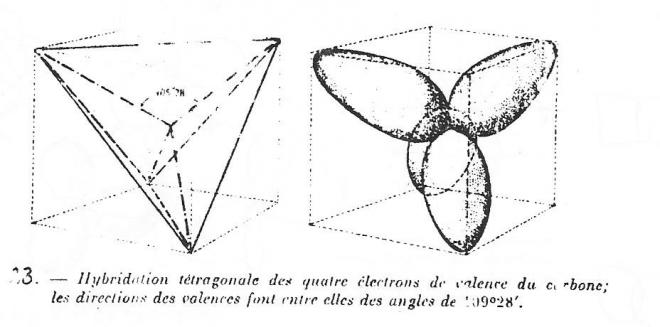 fig 8
fig 8
l'énergie entre deux liaisons quelconques est ici de 109°28'
Cette hybridation interviendra chaque fois que le carbone s'unit à 4 autres atomes, par exemple dans le méthane, l'éthane et les paraffines, les liaisons formées sont du type Sigma.
2° Hybridation sp2 ou trigonale
Il est nécessaire d'envisager ce nouveau type d'hybridation pour expliquer la formation d'une double liaison entre deux atomes de carbone ( composés éthyléniques ).
Dans ce cas , une des trois orbitales p reste intacte, les deux autres fusionnent avec l'orbitale s pour se réorganiser en trois orbitales hybrides. Celles-ci s'orientent avec un maximum de symétrie . Leurs axes, à 120 °les uns des autres , sont contenus dans le plan des orbitales hybridées, plan perpendiculaire à l'axe de l'orvitale p qui subsiste ( fig 9).
fig 9
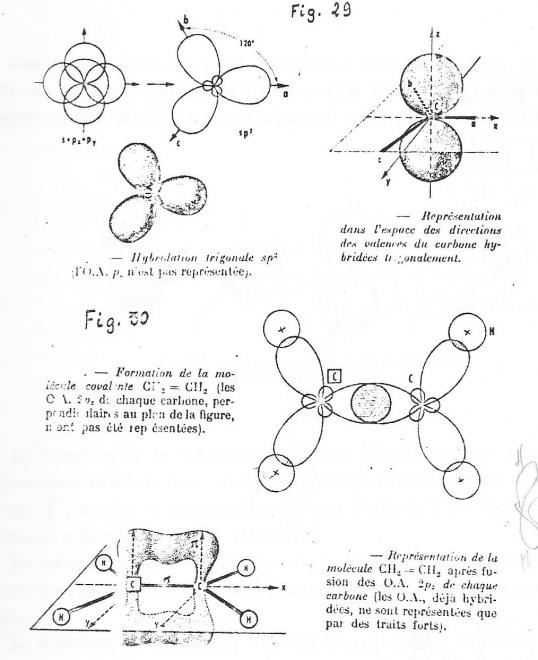 fig 10
fig 10
La formation de la double liaison C=C consiste alors en la superposition d'une liaison Sigma obtenue par recouvrement axial de deux orbitales hybrides et d'une liaison pi formée par recouvrement latéral des orbitales restées intactes .
La formation de l'écétylène résulte du recouvrement des deux orbitales hybrides vacantes sur chaque c par des orbitales atomiques d'atomes d'hydrogène
3° Hybridation sp1 ou digonale .
C'est le type d'hybridation rencontré dans le cas du béryllium. Elle est nécessaire à l'explication de la formation d'une liaison triple entre deux atomes de carbone ( dérivés acétyléniques ).
dans ce cas, deux des orbitales p restent intactes, la troisième disparaît avec l'orbitale s pour donner deux orbitales hybrides sp1 à symétrie linéaire, dans un plan perpendiculaire à celui des orbitales p intactes ( fig 11 ).
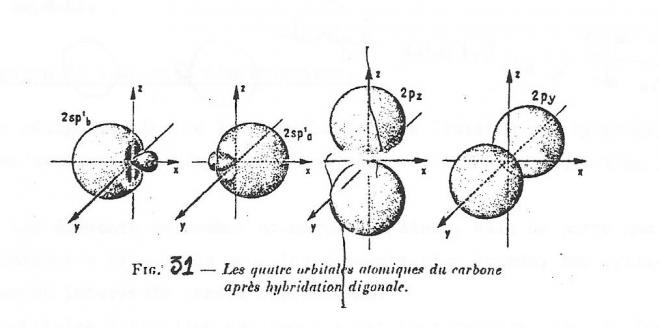 fig 11
fig 11
La formation de la molécule d'acétylène s'explique alors ainsi : d'abord, par recouvrement axial de deux orbitales hybrides formation d'une première liaison du type Sigma, suivie de la formation de deux liaisons pi par recouvrement latéral des orbitales p restées intactes ( fig 12 )
La molécule d'acétylène résulte de la formation de liaison Sigma supplémentaire des deux autres orbitales hybrides avec des orbitales atomiques de l'hydrogène.
La formation de liaison(s) pi permet d'expliquer le phénomène d'isomérie cis-trans dans le cas des dérivés éthyléniques.
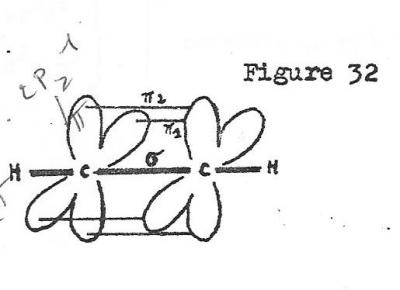 fig 12
fig 12
alors que , si deux carbones sont simplement liés par une liaison sigma , rien ne s'oppose à ce que le système des 3 liaisons restantes tourne autour de l'axe de cette liaison Sigma, la formation d'une liaison pi bloque le système, la libre rotation est empêchée.
c) Généralisation de la notion d'hybridation .
Cette réorganisation de la distribution des liaisons par hybridation ne s'applique pas seulement au béryllium et au carbone, mais à presque tous les éléments.
Pour les éléments de nomlbre atomique peu élevé, elle ne porte que sur les orbitales atomiques s et p, mais pour les éléments plus lourds, les orbitales d peuvent également intervenir dans l'hybridation.
Les orbitales d que l'on est amené à utiliser doivent être du même niveau quantique principal que les autres orbitales ou mieux de niveau inférieurs, ainsi que nous le verrons dans l'étude de la stabilité des complexes des métaux de transition.
Les orbitales hybrides obtenues s'orientent toujours avec un maximum de symétrie.
sp : linéaire
sp2 : triangle équilatéral
sp3 : tétraèdre
sp2d : carré ( Ni(CN)4)2-
sp3d : hexaèdre
sp3d2 d2sp3 : octaèdre ( SF6 )
Date de dernière mise à jour : 02/12/2018
Ajouter un commentaire